





During the course of evolution, multicellular organisms have been orchestrated with an efficient and versatile immune system to counteract diverse group of pathogenic organisms. Pathogen recognition is considered as the most critical step behind eliciting adequate immune response during an infection. Hitherto Toll-like receptors (TLRs), especially the surface ones viz. TLR2 and TLR4 have gained immense importance due to their extreme ability of identifying distinct molecular patterns from invading pathogens. These pattern recognition receptors (PRRs) not only act as innate sensor but also shape and bridge innate and adaptive immune responses. In addition, they also play a pivotal role in regulating the balance between Th1 and Th2 type of response essential for the survivability of the host. In this work, major achievements rather findings made on the typical signalling and immunopathological attributes of TLR2 and TLR4 mediated host response against the major infectious diseases have been reviewed. Infectious diseases like tuberculosis, trypanosomiasis, malaria, and filariasis are still posing myriad threat to mankind. Furthermore, increasing resistance of the causative organisms against available therapeutics is also an emerging problem. Thus, stimulation of host immune response with TLR2 and TLR4 agonist can be the option of choice to treat such diseases in future.
Antimicrobial inflammatory response primarily onsets through initial sensing of distinct pathogen associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) of hosts. These receptors serve as crucial innate PRRs that sense microbial or endogenous products released from damaged or dying cells and trigger innate immunity through the activation of intracellular signal transduction pathways.1 Amongst the innate immune PRRs, Toll-like receptors (TLRs) have the unique capacity to sense the initial infection and are the most potent inducers of the inflammatory responses.1 Depending on their cellular localization or respective PAMPs they identify, TLRs can be divided into two sub groups such as transmembrane (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) and intracellular (TLR3, TLR7, TLR8, and TLR9).1 These evolutionary conserved type-I transmembrane proteins (TLRs) can recognize ligand from almost all types of pathogenic organisms including viruses, bacteria, fungi, protozoa, helminths, etc. Structurally, TLRs located on cell membranes possesses an extracellular domain containing leucine-rich repeats that recognize distinct PAMPs and a toll-interleukin 1 (IL-1) receptor (TIR) domain required for downstream signalling that guides activation of transcription factor nuclear factor-κB (NF-κB) for inducing pro-inflammatory cytokines and chemokines as well as the up-regulation of co-stimulatory molecules on antigen presenting cells, such as macrophages (MΦs) and dendritic cells (DCs) that in turn sensitize T-cell activation. Inflammation signalled from TLR is a protective measure of the host body to ensure removal of detrimental threats posed by infectious agents as well as to accelerate the healing process. However, the Th1 biased inflammatory consequences orchestrated by TLRs not only involve in eliminating pathogenic infections but also can induce fatal pathological outcomes like septic shock (Fig. 1). Similarly, pathogen modulated TLR signalling develops a Th2 based response beneficial for the pathogen i.e. disease progression (Fig. 1). Thus, an adequate balance between pro- and anti-inflammatory immune responses is of immense importance to restore the normal physiological conditions of the host body during and after a pathogenic infection.2 Herein, major research findings exploring the role of TLR2 and TLR4 in the induction of host immunity against major parasitic diseases such as tuberculosis, leishmaniasis, malaria, trypanosomiasis, and filariasis have been reviewed.
Immunobiology of TLR2 and 4 or anti-inflammatory (Th2) polarization of immune cells.")
Since their discovery, TLR2 and 4 have gained much attention due to their extreme ability of identifying diversified array of pathogenic ligands.1 Alike Drosophila protein ‘Toll’, mammalian TLR2/4 possesses a cytosolic IL-1 receptor homolog domain but heterologous extracellular leucine-rich repeats.3 Interestingly, the mode of signalling is highly similar for e.g. transcription factor ‘Dorsal’ activated by Toll pathway in Drosophila is a functional homologue of NF-κB.4 The mode of activation of NF-κB or Dorsal also share high degree of similarity in terms of signalling intermediates like protein kinases such as ‘Pelle’ of Drosophila5,6 and mammalian IL-1 receptor-associated kinase.7 A comparative homology in the signal pathway transduced by Drosophila ‘Toll’ and mammalian “TLR” have been depicted in Fig. 2.
 and mammalian TLR pathway (right) in response to extracellular ligand (s).")
The functional features of “Toll signaling” are primarily different from insect to mammals. As obvious, ‘Toll’ receptors in insect (Drosophila) perform developmental roles primarily but it serves as innate immune receptor majorly in mammals. The proteins involved in the dorso-ventral polarity also play crucial role in the antimicrobial response in the fly as well.8 Interestingly, intermediates of dorso-ventral polarity determining ‘Toll’ pathway of insect share high degree of homology with vertebrate TLRs (specifically TLR4) that performs pattern recognition.8 Thus, the functional ancestry between development and immune pathway has been emerged as a major question. In particular, the discovery of immune function of ‘Toll’ in Drosophila also suggested towards the fact that the immune function of the Toll gene product was not adapted by the higher animals rather it can be revealed that the coordination and integration and/or cross talk between the intermediates of ‘Toll signaling’ has been improved with the increase in complexity among animals most likely during the course of evolution.8 In addition, unrelenting selective pressure exerted by the rapidly evolving pathogenic organism also contributed in this adaptive evolution of ‘Toll’ receptor.8 Particularly for TLR4, evolution of the gene (mostly due to mutation) led to differential expression of TLR4 with different affinity and specificity to its PAMP8 which may be the reason behind resistance/susceptibility to an infectious disease. However, the exact molecular explanation of the mechanism involved in the introduction of immune functioning in the “Toll pathway” alongside its developmental function or its functional divergence still remains as a shaded area in our understanding.
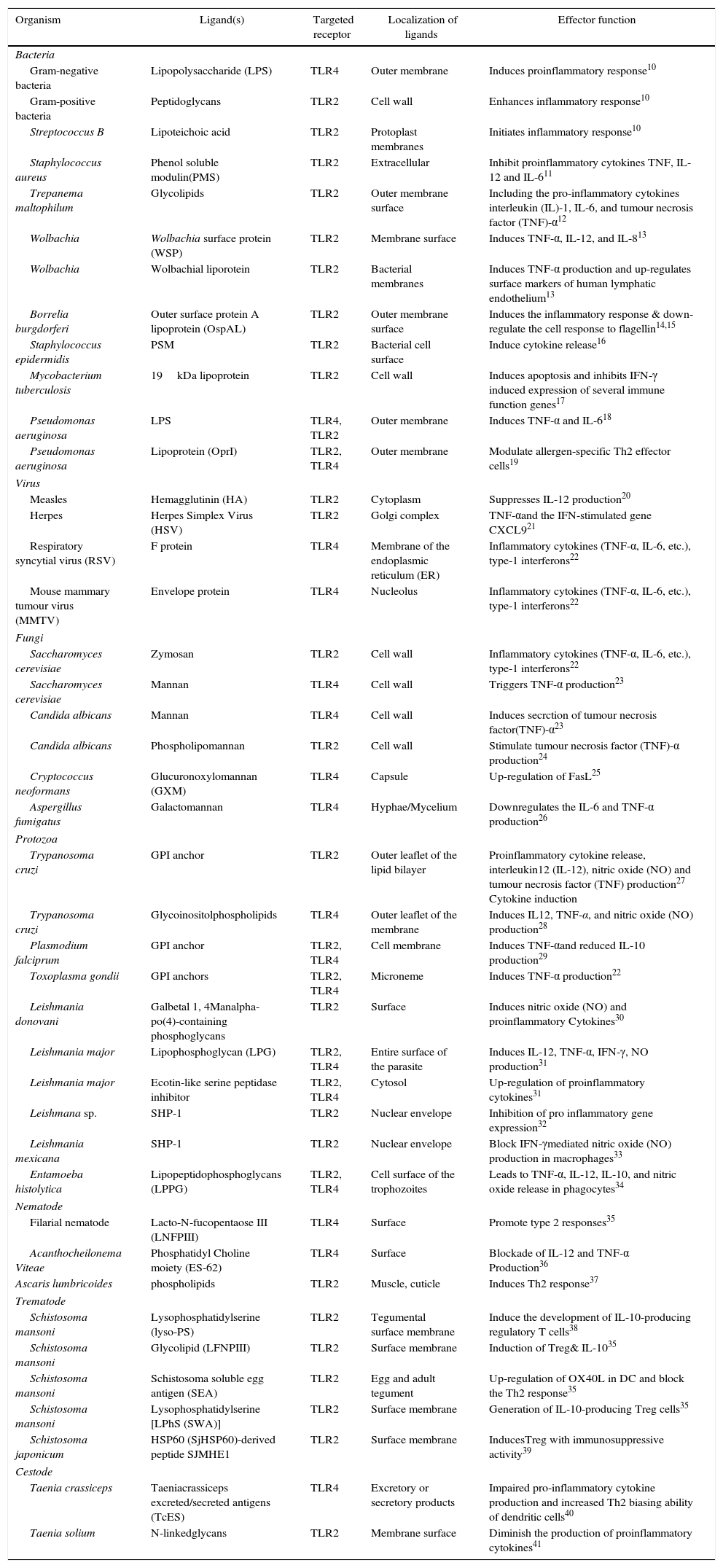
Being among the cell surface TLRs, TLR2 and 4 shape pathogen specific innate immunity through distinct ligand binding which in turn develops antigen-specific acquired immunity as well. These two TLRs are the best characterized PRRs which not only identify invading pathogens outside the cell but also intracellular pathogens captured in endosomes or lysosomes.9 TLR2 and 4 can sense PAMPs from various infectious micro and macro organisms as summarized in Table 1. The best possible explanation of cell surface TLR activation is the presentation and binding of lipopolysaccharide (LPS) to TLR4. Under pathogenic condition, a soluble plasma protein namely LPS-binding protein (LBP) interacts and binds with LPS.9 The entire LPS-LBP complex handed over to glycosyl phosphatidyl inositol linked CD14 firstly and then on to the TLR4-MD2 complex.9 TLR4 forms a complex with MD2 on the cell surface which serves as the main LPS-binding component.1 Five out of the six lipid chains of LPS occupy the hydrophobic pocket of MD2 and the remaining lipid chain exposed to the surface on MD2 associates with TLR4.1 The phosphate groups on sugar moieties also interact with the positively charged residues of TLR4. The multimeric receptor composed of two copies of the TLR4-MD2-LPS complex resulting initiation of signal transduction by recruiting intracellular adaptor molecules such as Myeloid differentiation factor 88 (MyD88), TIR-related adaptor protein inducing interferon (TRIF), TRIF-related adaptor molecule (TRAM), TIR domain containing adaptor protein (TIRAP) or MyD88 adaptor like (MAL), and Sterile-alpha and Armadillo motif-containing protein (SARM). The summary of mode of ligand recognition by TLR4 has been depicted in Fig. 3.
Recognition of ligand and functional immunobiology of different TLRs against array of pathogens.
| Organism | Ligand(s) | Targeted receptor | Localization of ligands | Effector function |
|---|---|---|---|---|
| Bacteria | ||||
| Gram-negative bacteria | Lipopolysaccharide (LPS) | TLR4 | Outer membrane | Induces proinflammatory response10 |
| Gram-positive bacteria | Peptidoglycans | TLR2 | Cell wall | Enhances inflammatory response10 |
| Streptococcus B | Lipoteichoic acid | TLR2 | Protoplast membranes | Initiates inflammatory response10 |
| Staphylococcus aureus | Phenol soluble modulin(PMS) | TLR2 | Extracellular | Inhibit proinflammatory cytokines TNF, IL-12 and IL-611 |
| Trepanema maltophilum | Glycolipids | TLR2 | Outer membrane surface | Including the pro-inflammatory cytokines interleukin (IL)-1, IL-6, and tumour necrosis factor (TNF)-α12 |
| Wolbachia | Wolbachia surface protein (WSP) | TLR2 | Membrane surface | Induces TNF-α, IL-12, and IL-813 |
| Wolbachia | Wolbachial liporotein | TLR2 | Bacterial membranes | Induces TNF-α production and up-regulates surface markers of human lymphatic endothelium13 |
| Borrelia burgdorferi | Outer surface protein A lipoprotein (OspAL) | TLR2 | Outer membrane surface | Induces the inflammatory response & down-regulate the cell response to flagellin14,15 |
| Staphylococcus epidermidis | PSM | TLR2 | Bacterial cell surface | Induce cytokine release16 |
| Mycobacterium tuberculosis | 19kDa lipoprotein | TLR2 | Cell wall | Induces apoptosis and inhibits IFN-γ induced expression of several immune function genes17 |
| Pseudomonas aeruginosa | LPS | TLR4, TLR2 | Outer membrane | Induces TNF-α and IL-618 |
| Pseudomonas aeruginosa | Lipoprotein (OprI) | TLR2, TLR4 | Outer membrane | Modulate allergen-specific Th2 effector cells19 |
| Virus | ||||
| Measles | Hemagglutinin (HA) | TLR2 | Cytoplasm | Suppresses IL-12 production20 |
| Herpes | Herpes Simplex Virus (HSV) | TLR2 | Golgi complex | TNF-αand the IFN-stimulated gene CXCL921 |
| Respiratory syncytial virus (RSV) | F protein | TLR4 | Membrane of the endoplasmic reticulum (ER) | Inflammatory cytokines (TNF-α, IL-6, etc.), type-1 interferons22 |
| Mouse mammary tumour virus (MMTV) | Envelope protein | TLR4 | Nucleolus | Inflammatory cytokines (TNF-α, IL-6, etc.), type-1 interferons22 |
| Fungi | ||||
| Saccharomyces cerevisiae | Zymosan | TLR2 | Cell wall | Inflammatory cytokines (TNF-α, IL-6, etc.), type-1 interferons22 |
| Saccharomyces cerevisiae | Mannan | TLR4 | Cell wall | Triggers TNF-α production23 |
| Candida albicans | Mannan | TLR4 | Cell wall | Induces secrction of tumour necrosis factor(TNF)-α23 |
| Candida albicans | Phospholipomannan | TLR2 | Cell wall | Stimulate tumour necrosis factor (TNF)-α production24 |
| Cryptococcus neoformans | Glucuronoxylomannan (GXM) | TLR4 | Capsule | Up-regulation of FasL25 |
| Aspergillus fumigatus | Galactomannan | TLR4 | Hyphae/Mycelium | Downregulates the IL-6 and TNF-α production26 |
| Protozoa | ||||
| Trypanosoma cruzi | GPI anchor | TLR2 | Outer leaflet of the lipid bilayer | Proinflammatory cytokine release, interleukin12 (IL-12), nitric oxide (NO) and tumour necrosis factor (TNF) production27 Cytokine induction |
| Trypanosoma cruzi | Glycoinositolphospholipids | TLR4 | Outer leaflet of the membrane | Induces IL12, TNF-α, and nitric oxide (NO) production28 |
| Plasmodium falciprum | GPI anchor | TLR2, TLR4 | Cell membrane | Induces TNF-αand reduced IL-10 production29 |
| Toxoplasma gondii | GPI anchors | TLR2, TLR4 | Microneme | Induces TNF-α production22 |
| Leishmania donovani | Galbetal 1, 4Manalpha-po(4)-containing phosphoglycans | TLR2 | Surface | Induces nitric oxide (NO) and proinflammatory Cytokines30 |
| Leishmania major | Lipophosphoglycan (LPG) | TLR2, TLR4 | Entire surface of the parasite | Induces IL-12, TNF-α, IFN-γ, NO production31 |
| Leishmania major | Ecotin-like serine peptidase inhibitor | TLR2, TLR4 | Cytosol | Up-regulation of proinflammatory cytokines31 |
| Leishmana sp. | SHP-1 | TLR2 | Nuclear envelope | Inhibition of pro inflammatory gene expression32 |
| Leishmania mexicana | SHP-1 | TLR2 | Nuclear envelope | Block IFN-γmediated nitric oxide (NO) production in macrophages33 |
| Entamoeba histolytica | Lipopeptidophosphoglycans (LPPG) | TLR2, TLR4 | Cell surface of the trophozoites | Leads to TNF-α, IL-12, IL-10, and nitric oxide release in phagocytes34 |
| Nematode | ||||
| Filarial nematode | Lacto-N-fucopentaose III (LNFPIII) | TLR4 | Surface | Promote type 2 responses35 |
| Acanthocheilonema Viteae | Phosphatidyl Choline moiety (ES-62) | TLR4 | Surface | Blockade of IL-12 and TNF-α Production36 |
| Ascaris lumbricoides | phospholipids | TLR2 | Muscle, cuticle | Induces Th2 response37 |
| Trematode | ||||
| Schistosoma mansoni | Lysophosphatidylserine (lyso-PS) | TLR2 | Tegumental surface membrane | Induce the development of IL-10-producing regulatory T cells38 |
| Schistosoma mansoni | Glycolipid (LFNPIII) | TLR2 | Surface membrane | Induction of Treg& IL-1035 |
| Schistosoma mansoni | Schistosoma soluble egg antigen (SEA) | TLR2 | Egg and adult tegument | Up-regulation of OX40L in DC and block the Th2 response35 |
| Schistosoma mansoni | Lysophosphatidylserine [LPhS (SWA)] | TLR2 | Surface membrane | Generation of IL-10-producing Treg cells35 |
| Schistosoma japonicum | HSP60 (SjHSP60)-derived peptide SJMHE1 | TLR2 | Surface membrane | InducesTreg with immunosuppressive activity39 |
| Cestode | ||||
| Taenia crassiceps | Taeniacrassiceps excreted/secreted antigens (TcES) | TLR4 | Excretory or secretory products | Impaired pro-inflammatory cytokine production and increased Th2 biasing ability of dendritic cells40 |
| Taenia solium | N-linkedglycans | TLR2 | Membrane surface | Diminish the production of proinflammatory cytokines41 |
 to TLR4 through the coordinated actions of serum LBP, membrane bound CD14 and MD2.")
TLR2 forms an m-shaped heterodimers with either TLR1 or TLR6 for recognizing tri- or di-acylated lipopeptides respectively from the pathogens (Gram-negative bacteria, mycoplasma, etc.).3 Out of the three lipid chains of tri-acylated ligand (lipopeptide e.g. Pam3CSK4), two interact with TLR2 in the TLR2-TLR1 heterodimer whereas the third chain occupies the hydrophobic channel of TLR1.7 An absence of hydrophobic channel in TLR6, TLR2-TLR6 heterodimer cannot recognize triacylated lipopeptides. Other than discrimination between lipoproteins, TLR2 has the ability to act together with co-receptor (CD36) by working with the heterodimer to mediate the sensing of some but not all TLR2 agonists which induces internalization.42 Thus TLR2 is involved in the recognition of a wide range of PAMPs derived from bacteria, fungi, parasites and viruses also. TLR2, together with its co-receptors, TLR1 and TLR6, requires internalization to trigger NF-κB activation in response to Lipoteichoic acid (LTA) and Pam3CSK4 that provides a novel accepting mechanism on how TLRs coordinate ligand recognition and subsequent triggering of a specific signal.43 Despite the expression of TLR2 at the cell surface, the bacterial TLR2 ligands namely Pam3CSK4 and LTA can induce NF-κB-dependent signalling from endosomal compartments reported for human monocytes and for a NF-κB sensitive reporter cell line.43
TLR2 and 4 are expressed in various immune cells including neutrophils, monocytes/MΦs and DCs.44 Amongst these, neutrophils first migrate to the site of infection, sense the pathogen and elicit immune response. However, coordinated activation of adaptive response is mediated through binding of specific ligand to monocytes or DCs that are also mediated principally by TLR2 and 4.45 Moreover, these TLRs are also expressed on classical adaptive immune cells viz. B and T lymphocytes.46
After recognizing PAMPs by the TLR2 and 4 in a particular cell type, cytosolic TIR domain and associated adaptors (MyD88, TRIF, and SARM) are activated that in turn drive activation of the transcription factors such as NF-κB and/or IRF3. Activation of these TLRs triggers two distinct signalling pathways viz. MyD88 dependent and MyD88 independent or TRIF dependent pathway. MyD88, TRIF, TRAM, MAL, and SARM are the five important adaptors of TLR activation. MyD88 is the first amongst the adaptors used by all TLRs except TLR3. Upon TLR activation, MyD88 recruits interleukin 1 receptor associated kinase 4 (IRAK-4) that phosphorylates IRAK1 which in turn activate TNF Receptor-Associated Factor 6 (TRAF6). Both proteins leave the receptor complex and interact with TGF-β-activated kinase 1 (TAK1) and two TAK1 binding proteins TAB1 and TAB2. TAK1 becomes phosphorylated and activates the I-κB kinase (IKK) complex comprising IKKα, IKKβ, and NEMO/IKKγ, and Mitogen-activated protein kinase (MAPK) leading to the activation of NF-κB and c-Jun N-terminal kinases (JNK) signalling pathways respectively for triggering the expression of cytokines (IL-6, IL-12, TNF-α).47 I-κB is then phosphorylated allowing NF-κB translocation to the nucleus and induces expression of pro-inflammatory cytokines. TAK1 is also capable of phosphorylating MKK6 and 7 which leads to the activation of p38 and JNK. MAL or TIRAP is the second adaptor that transmits signal from TLR4 and TLR2 by facilitating movement of MyD88 to TLR4 and/or possibly to TLR2 for transcription factor activation like NF-κB, JNK and Extracellular regulated kinases 1(ERK-1).48 TRIF is the third adaptor for TLR4 and TLR3 that activates Interferon regulatory factor 3 (IRF3) by activating the kinases TBK1 and the I-κB kinase €. Fourth adaptor is TRAM which is one of the interacting partners of TRIF. TRAM involves in transmitting TLR4 signalling and results in MyD88-independent interferon-β production.49 The fifth adaptor is SARM which mainly interact with TRIF and negatively regulate NF-κB and IRF3 activation.50
Activation of IRF3 and NF-κB are the principal targets of TRIF-dependent pathway.51 TRIF recruits TRAF6 and activates TAK1 for NF-κB activation through ubiquitination dependent mechanisms similar to those of the MyD88 dependent pathway.45 TRIF forms a multiprotein signalling complex along with TRAF6, TNFR-associated death domain (TRADD), Pellino-1 and Receptor-interacting protein1 (RIP1) required for the activation of TAK1, which further activates NF-κB and MAPK.1 In addition to NF-κB activation, the TRIF-dependent pathway also leads to IRF3 activation and interferon-β transcription.52 TRIF recruits another signalling complex involving the non-canonical IKKs, TBK1 and IKKi (IKK¿), which catalyze the phosphorylation of IRF3 and induce nuclear translocation to mediate transcription of target genes.53 TRAF3 is also incorporated into the MyD88 complex during signalling. Degradation of TRAF3 results in the translocation of the membrane proximal signalling complex to the cytoplasm leading to TAK1 activation that phosphorylates the IKK complex to mediate NF-κB activation.54 The mechanism of signalling induced from TLR-ligand interface to mediate inflammatory response has been presented in Fig. 4.
Involvement of TLR2 and TLR4 in infectious diseases
Diseases caused by parasites/microbes are just not the burden for mankind; they are economically considered as biohazards. Major infectious diseases like tuberculosis (TB), malaria, leishmaniasis, trypanosomiasis, and filariasis are the deadliest for the extent of harm they result. In this section, current scenario of the diseases of our interests has been presented with up-to-date information.
TuberculosisTuberculosis (TB) caused by the intracellular bacterium Mycobacterium tuberculosis, capable of surviving within host mononuclear cells, remains one of the world's deadliest communicable diseases. TB is a major public health problem causing 10 million new infected cases diagnosed each year whereas deaths of 2 million victims.55 In 2013, an estimated 9.0 million people developed TB and 1.5 million died from the disease worldwide (WHO, 2014). TLR2 and 4 significantly contribute in the typical immune response in TB and their interaction with Mycobacterium tuberculosis believed to be the principal event. The decision behind progression or elimination depends what type of microbacterial ligand binds to these receptors.56 In general, mycobacteria have adopted sophisticated evasion strategy to bypass MΦs through alternative activation whereas elimination of mycobacterial infection is predominantly occurring by classically activated MΦs.57 Kusner58 has reported a 19kDa mycobacterial secretary lipoprotein that activates murine and human MΦs to secrete TNF-α and nitric oxide (NO) via interaction with TLR2, but not TLR4. Mycobacterial glycolipids like lipoarabinomannan (LAM), lipomannans (LM), phosphatidylinositol mannosides (PIM2, PIM6), and a 19-kDa lipoprotein, are the mycobacterial PAMPs believed to cause MΦ activation via the TLR2 activating innate pathway by regulating tumour necrosis factor (TNF-α) and IL-10 secretion reported for human monocyte-derived MΦs.57 The interaction of MΦs with M. tuberculosis through TLRs is critical in defining the cytokine profile that may or may not control disease progression. This interaction promotes the synthesis of antibacterial molecules (e.g., NO) and several cytokines, including TNF-α, interleukin-6 (IL-6), IL-1β, and IL-12.57 In DCs, the engagement of TLR2 with these PAMPs induces the secretion of anti-inflammatory cytokines such as IL-10, a cytokine that promotes disease progression, by mechanisms that limit the inflammatory response.57 Along with cytokine expression TLR2 activation leads to NO dependent and independent killing of intracellular M. tuberculosis in mouse and human MΦs respectively.59 Apoptosis by infected host cells is also mediated by TLR2 mediated pathway involving MyD88, Fas-Associated protein with Death Domain (FADD) and caspase-8 in which MyD88 associates with FADD via their respective death domains.60 A scheme on TB induced TLR activation has been shown in Fig. 5.
Malaria
Malaria is a mosquito borne infectious disease of humans and other animals caused by parasitic protozoan of genus Plasmodium (WHO, 2014) causing huge number of death each year.61 The host response during malaria infection, expresses high levels of many proinflammatory cytokines, including TNF-α, IL-1, IL-6, IL-12 and IFN-γ that have important roles in controlling parasite growth.62 Such types of responses are primarily originated from interaction of malarial surface molecules and TLRs of host immune cells. Till date, search for TLR activating PAMPs from malarial parasites have been the major highlighted objective for last few decades. In this connection, a glycolipid moiety, glycosyl phosphatidyl inositol (GPI) from P. falciparum has been reported to be a ligand of both TLR2 (heterodimeric form with TLR1 and TLR6) and TLR4.63 Malarial GPI is structurally dissimilar than human and comprises a conserved ethanolamine phosphate-substituted oligosaccharide moiety, EtN-P-6Mana1-2Mana1-6Mana1-4GlcN, linked to phosphatidylinositol by a (1-6)-glycosidic bond.29 The covalently linked fatty-acyl moieties of GPIs display a specific pattern for recognition by TLR2 and the co-receptors. This malarial ligand from infected erythrocytes interact with the mentioned TLRs located on MΦs or DCs and contribute to the immunopathology of the infection by inducing the expression of proinflammatory cytokines, such as TNF-α, IL-6 and IL-1. In addition, GPI anchored proteins, merozoite surface protein 1 (MSP1) and MSP2, two major asexual blood stage surface antigens, have endotoxin activity that indeed results in decreasing frequency of the parasitic load from host.63 Moreover, such immunostimulatory host response by these surface proteins are induced through their binding to TLR4 and activation of immune signalling leading to release of cytokines from MΦs or DCs. P. falciparum GPI-induced MΦ activation is typically CD36 dependent transmits through MAPK, namely extracellular signal regulated kinase (ERK), p38 and c-Jun N-terminal kinase (JNK) pathways as well as through the Nuclear factor- kappa B (NF-κB).29 In addition, studies using inhibitors of p38 activity and ERK1/2 activation indicated towards significant crosstalk among the GPI activated MAPK pathways represents redundant and overlapping functions in the initiation and regulation of innate immune responses.29 Activation of TLR4/MyD88 dependent signalling pathway during the malarial infection results in the excessive proinflammatory cytokine production responsible for the several symptoms of the diseases.29 This adaptor protein plays a more prominent role in pathogenesis, rather than in protection. A scheme on malaria induced TLR activation has been shown in Fig. 5.
In particular, reactive oxygen and nitrogen radicals produced by the malaria sensitized MΦs guides immune system by TNF-α, IL-12, and IFN-γ to kill these parasites through the induction of apoptosis.29 These proinflammatory cytokines facilitate parasite-specific adaptive immunity for efficient parasite clearance to sustained and effective protection against disease pathology. However, overwhelmed proinflammatory response often results in fatal outcomes.29 On other side, cell-mediated adaptive immune responses and the modulation of humoral responses by the induction of antibody isotype switching occur mainly by the involvement of IL-12.29 Generally, a gradual down-regulation of proinflammatory cytokine production occurs with a parallel increase in the expression of anti-inflammatory cytokines (IL-10) when the parasite burden starts declining during malaria infection.29 However this balance is required for fighting against the infection while escaping pathogenesis might not always be reached, contributing to medical situations.
LeishmaniasisLeishmaniasis is caused by a protozoa parasite and transmitted to humans by the bite of infected female phlebotomus sandflies. This disease is the second-largest parasitic killer in the world (after malaria), responsible for an estimated 200,000–400,000 infections each year worldwide (WHO, 2015). The anti-leishmanial host response typically depends on the quality of the adaptive immune response primarily induced from host-parasite interaction. Especially, interaction between leishmanial parasite and innate immune receptors rather TLRs results generation of inflammatory mediators to deprive parasite burden and develops efficient adaptive responses. Lipophosphoglycan (LPG), the major identified ligand from these parasites, has been reported to induce inflammation through TLR2 and 4.64,65 LPG occurs both in the parasite surface as well as in the secretory form and these two forms are structurally similar but differ in the average number of phosphorylated oligosaccharide repeat units present and in the sugar types present in the glycan. Up-regulation and activation of TLR2 on human Natural killer (NK) cells by LPG of L. major results enhancement of TNF-α and IFN-γ. However, interaction of LPGs with the TLRs is also influenced by the structural diversity of the phosphoglycan. Likewise, LPG of L. major, L. mexicana, L. aethiopica, and L. tropica were reported as classical TLR2 ligand of MΦs whereas performance of L. tropica LPG is not so much satisfactory.65 Study on the induction of TNF-α in MΦ by L. major LPG showed need of a lipid anchor and a functional MyD88 adaptor.65 Such MyD88-dependent pathways are particularly important in developing protective IL-12-mediated Th1 response against the parasite.65 The L. infantum protein related to the silent information regulator 2 (SIR2) family has been reported for stimulating the proliferation of activated B lymphocytes, overexpression of major histocompatibility complex- (MHC) II and the co-stimulatory molecules CD40 and CD86 as well as maturation of DCs accompanied by the secretion of IL-12 and TNF-α are principally governed through TLR2.64 In spite of its proinflammatory activity, LPG has been shown to inhibit the progression of inflammatory signalling originated from TLR2/TLR4 by inducing the expression of suppressors of the cytokine signalling (SOCS) family proteins viz. SOCS-1 and SOCS-3.65,66 Another exciting fact about LPG is that production of ROS is induced through the interaction between membrane LPG and immune cell TLR-2 leading to the differentiation of Th2 subsets whereas Th1-promoting cytokines are reported to be induced solely by soluble LPG.65 LPG potentiated inflammatory responses through TLR2 stimulation and IL-12 expression followed by Th1 responses in mice was also reported after treating with synthetic oligosaccharide analogues of LPG glycan structure indicating the importance of glycan in the immunostimulatory activity of LPG.65 Interestingly, parasites also use the same ligand as well as the TLRs to supress the immune cell activation required for parasites’ benefit i.e. disease progression. Faria et al.65 reported L. donovani infected human THP1-derived MΦs showed suppression in the TLR2 stimulated IL-12 release with increased production of IL-10 resulted from the inhibition of MAPKp38 phosphorylation and activating ERK1/2 phosphorylation. In addition, L. donovani, L. mexicana, and L. major have been reported to exploit the MΦ tyrosine phosphatase SHP-1 to inactivate kinases (e.g. IRAK) involved in TLR signalling.32 A graphical summary on leishmanial parasite induced TLR activation is shown in Fig. 5.
Proteoglycolipid complex (P8) composed of acysteine and serine metalloprotease, host-derived Apolipoprotein E (ApoE), and four glycolipids has been reported as another potential TLR4 ligand of Leishmania detected in L. pifanoi amastigotes that influence phagocytosis through TLR activation.65 Activated TLR4 at the parasitophorous vacuole affects the microenvironment surrounding the parasite for its control. Particularly for the neutrophils, TLR4 induces IRAK4 for exocytosis of neutrophil secretory granules.65 Mechanistically, P8 sensitized MΦs induce proinflammatory cytokines viz. IL-1β and TNF-α through TLR4, MD2, CD14, and MyD88.65 In addition, metacyclic promastigotes have been reported to induce phosphorylation of the MAP kinases such as ERK, p38, and JNK through TLR4 in L. mexicana.67 During such course of inflammation of MΦ, iNOS, Cyclooxygenase-2 (COX-2), Prostaglandin E2 (PGE2), NO and arginase-1 are been the mediators.67
Trypanosomiasis (Chagas disease)Chagas disease or American trypanosomiasis, is a tropical parasitic disease caused by the protozoan Trypanosoma cruzi (CDC, 2015) and transmitted by insects vector like Triatominae or kissing bugs. In Latin America it causes a health threat for an estimated 10 million people and more than 25 million people are at risk. Typical immunity against Chagas disease is principally mediated by innate recognition through TLRs. Although studied less, TLRs have been characterized as crucial determinants in evoking anti- T. cruzi immune response in which both TLR2 and TLR4 are the key mediators.68 In particular, these receptors mediate internalization of trypanosomatid through phagocytosis required for initiating the immune response by phagosomal maturation to kill the parasite followed by antigen presentation.69 Researchers have demonstrated fusion of early endosomes, and phagocytosis induced by trypomastigotes in MΦs by ligation of guanine phosphonucleotide- binding proteins Ras-related protein- (Rab-) 5 solely through TLR2.69 TLR2 activation has been reported to proceed via MyD88-dependent pathway.69 Secretion of chemokines signalled from TLR2 activation further recruit leukocytes to control the infection in a broad way.28 In particular, TLR2 signalling is targeted by the trypanosomatids to hinder the immune response induced by the parasite.
However, TLR activation resulted from immune cells-T. cruzi interaction may also contribute to the disease pathology as well.69 Involvement of TLR activation in the immunopathology of chronic Chagas’ cardiopathy has been reported to be due to a single-nucleotide polymorphism in the genes coding for the signalling proteins of TLR signalling and leads to differential susceptibility to Chagas disease.69 For example, T. cruzi-infected individuals have heterozygous MAL/TIRAP S180L variant shows poorer signal transduction after binding of the trypanosomal ligand to TLR2 or TLR4 resulting in lesser chance of developing chronic Chagas’ cardiomyopathy.69 A summary on trypanosomatid induced TLR activation has been shown in Fig. 5.
FilariasisLymphatic filariasis, a vector borne neglected tropical disease, mainly caused by three species of nematode parasites- Wuchereria bancrofti, Brugia malayi, and B. timori is considered to be a major public health burden (WHO, 2015). Pathological manifestations of this disease mainly include lymphedema, elephantiasis and rarely a greater degree of neurological manifestation.70 Recent studies also showed existence of co-infection of filarial parasite with bacteria (TB),49 malaria 49,71 and opportunistic yeast.72 In general, the immunoresponse in filariasis is complicated and unresolved especially the involvement and functional role played by TLR2 and TLR4.49 Filarial intracellular symbiotic bacterium, Wolbachia has been reported to cause inflammation through its lipopolysaccharide (of outer membrane) after binding with TLR4. TLR2 and TLR4 are also capable to bind other Wolbachial products like Wolbachia surface protein (WSP) and induce inflammatory response by secreting pro-inflammatory cytokines from MΦs and DCs.13 Such proinflammatory responses are of immense importance to eliminate the infection from host. However, filarial nematodes also target TLR2 and TLR4, especially TLR4, for modulating host immune response. A phosphorylcholine- conjugated glycoprotein molecule namely ES-62 binds TLR4 and create anti-inflammatory/Th2 biased immune response to prolong survivability of the parasite inside host.36 In chronic infection, filarial nematode induces apoptotic death of T cells for down regulating host response and interestingly the process is also operative through TLR4.73 Live microfilarae of B. malayi have been reported to modulate TLR4 expression in monocyte derived human DCs (mhDCs) to induce an immunosuppressive environment in host through interference of MyD88-dependent suppression of NF-κB signalling to hinder production of proinflammatory cytokines and type-1 interferons. In addition to such inhibitory effect of microfilarae over DCs, these also results partial inhibition of the function of human Langerhans cells (LCs) and prevent proliferation of CD4+ T cells.74 Interestingly, TLR2 and TLR4 have been reported to be expressed on circulating B cells during helminthic infection that uphold and maintain Th2 type-immune responses to make “worm favourable” conditions.75 These specialized B cell subsets are known as regulatory B cells (Breg) and their immunosuppressive function is also regulated through TLR4.75 TLR4 on these cells trigger induction of IL-10 secretion from B cell through Myd88-dependent pathway.76 Particularly in filariasis, Breg cells are responsible for the induction as well as maintenance of hypo-responsive immune status with elevated levels of Treg, IL-10 and filarial-specific IgG4.75 This Breg mediated responses is recompensing as patients remain asymptomatic due to devoid of excessive immunopathology.75 However, this typical Th2 environment by Breg results secondary infections by bacteria and/or fungi due to lack of protective immune cells repertoires in filarial subjects.75 A probable mechanism of TLR activation by filarial parasites has been shown in Fig. 5.
Conclusion and future prospectsStudies on the recognition of infectious pathogens through TLRs have significantly contributed to our understanding of the underlying mechanisms of innate immune defense against the deadliest parasites. In this review, we have put forward the findings made on the role played by two crucial members of the TLR family viz. TLR2 and TLR4 against the most threatening infectious diseases. We have discussed different previously reported findings on types of pathogenic ligand alongside the mechanism of action of inflammatory cascade originated from TLR4 and TLR2 in tuberculosis, malaria, leishmaniasis, filariasis, and trypanosomiasis. Interesting outcomes achieved in the experimental vaccine adjuvants and/or immunotherapy against active leishmaniasis promises for effective future applications as well as opens new possibilities regarding the use of TLR based immunotherapeutic control strategies for malaria or other diseases. Similarly, immunomodulatory TLR4 ligand from filarial parasites has also shown to control allergic reaction by inhibiting proinflammatory mediators and this strategy could be used effectively to combat inflammatory pathology of malaria or other disease to reduce mortality. Similarly, TLR4 has been emerged as a target of therapeutics for immunopharmacological control of infectious and/or inflammatory diseases.77
However, many questions remain to be answered for this aspect. Thus more concluding information rather inferences are need to be gathered through population based studies. Similarly, there is a need to search for other microbial ligands for a same microbe/parasite as well as interplay between the TLRs and obviously the signalling crosstalk. From immunological perspectives, study of mechanism of parasite/microbe induced polarization of major antigen presenting cells may offer us new target for developing therapeutic strategy to restore Th1 (to eliminate infection) or Th2 biased (to protect host from overt inflammation) immune response.
Conflicts of interestThe authors declare no conflicts of interest.
SM acknowledges University Grants Commission for the award of his Senior Research Fellowship.
