
The formula proposed by Rich in 1951 explained the formation in a tuberculous lesion in a period that was unknown cellular functions, cytokines and other immunological aspects involved in granuloma formation by tuberculosis; its components are assembled conceptually to explain the pathogenic mechanisms involved in the granulomatous lesion in tuberculosis. In this manuscript, we report an update of Rich's formula based on the new and old concepts about pathogenic mechanisms involved in the granulomatous lesion in tuberculosis. Current knowledge allows us to conclude that the balance between the characteristics of the bacillus and host protective response is necessary to indicate the outcome of pathogenesis, infection or active disease and the necrosis degree of the tuberculosis lesion.
Tuberculosis (TB) is the most frequent cause of granulomas and is the most prevalent infectious disease in Brazil, remaining one the most important problems of public health in the world. In 2010, 71,000 cases were reported, with a mortality of 4800/year.1 Brazil is the 19th in rank among countries with the majority of TB cases in the world. Bahia, the northeast State of Brazil, is ranked 3rd in number of cases, after São Paulo and Rio de Janeiro. TB incidence in Bahia is 39.5/100,000 habitants.1–3
TB is a granulomatous disease characterized by a cell-mediated hypersensitivity reaction in response to components of the wall structure of Mycobacterium tuberculosis especially cord factor and lipoarabinomannan.4 Granuloma was first described in 1679 by Sylvius, and etymologically comes from the original word tubercle.5 Nowadays, granulomas are much better understood based on the knowledge of cellular immunology (cell mediated hypersensitivity reaction) and its interaction with cytokines and chemokines.
In 1951, Rich proposed that the pathogenesis of tuberculous lesion was directly dependent of inoculum, bacillus number (N), bacilli virulence (V) and host hypersensitivity (H), by counterbalancing and decreasing the lesion size, the natural and acquired resistance of the host (Rn+a). This formula can be represented by6:
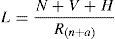
Rich's formula shows components that are assembled conceptually to explain the pathogenic mechanisms involved in the granulomatous lesion in TB. Considering the TB pathogenesis in two different clinical forms, infection and disease, where in infection, the balance depends on the host natural resistance. In contrast, in disease, the balance depends on the bacilli properties such as virulence and hypersensitivity phenomenon. In both clinical forms, either infection or disease, granulomas are detected in the tissue.7–9 Both stages are taking place inside of the granuloma, where the components of Rich's formula occur. The ancient concept was that a granuloma was a cellular agglomerate walling of the bacilli; thus in Rich's formula this should be considered in the denominator. This can be observed in some host, in which the granuloma formation is impaired and therefore the bacillus growth is faster. In fact, in these almost 60 years since the proposition of Rich's formula, the science has advanced significantly in the knowledge of the properties of M. tuberculosis, as well as in understanding of host defense mechanisms. These new concepts have been incorporated into the above formula.
Initiation of infection and granuloma formationAfter mycobacterial arrival in the tissues of the respiratory tract, an unspecified reaction occurs in which neutrophils try to phagocytize the bacillus. This initial phase is not well studied in humans.10 Three weeks later, there is a development within these tissues of a more specific reaction, when mononuclear cells reach the focus of the infectious lesion, modulating the granuloma formation.9,11,12 Mononuclear cells including monocytes/macrophages and lymphocytes, cells with their predefined morphologies and functions, participate in the recognition and presentation of components of M. tuberculosis to the immune system.11–13 During and after mononuclear cells naïve starts cells differentiation into specific cell types such as macrophages, dendritic cells, CD4 and CD8 T helper.14
In the process of granuloma formation, many macrophages with large cytoplasm and organelles with high phagocytic and microbicidal functions were observed.15–17 These cells can fuse into multinucleated giant cells; however, these changes are not completely understood.12,17 Mature macrophages can undergo a transformation into epithelioid cells (epithelioid histiocytes), which interdigitated their membranes aggregating into other cells as the epithelial barrier.16 CD4 and CD8 T helper (Th) lymphocytes as well as epithelioid monocytes and giant multinucleated cells participate in this granuloma formation that is orchestrated by INF-γ produced by CD4 and TNF-α secreted by macrophages.8,11,12,15,18 Other cells, such as neutrophils, join this granuloma via a dynamic process.9,19 All these facts have dynamic morphology that is influenced by the balance between host resistance and the M. tuberculosis virulence gene expression.9,11
Cellular apoptosis of caseous necrosis can also be detected within the centers of granulomas. Progress has been made in determining these mechanisms; however, much remains not completely understood.9 One interesting point that has been found regards the function of the granuloma. Although the main consensus is that the granuloma that surrounds the M. tuberculosis is doing so in an attempt to restrict it from spreading.20 Other experiments in the zebrafish have shown that granuloma also contribute to bacilli spreading. During the granuloma's formation the tissues are injured and necrosis develops. This mechanism of lesion development depends on the acquired resistance versus the hypersensitivity developed by the patient to the bacillus during the infection.9
Granulomas seen in tuberculous pleurisy allow to distinguish the cellular components comparing the cells present in pleura fluid.21 Fayyazi et al. showed a possible apoptosis of epithelioid cells and the presence of CD4 T lymphocytes in the proximity of the focus of the necrosis. Such findings could suggest a direct relation between the cells, their secretions and the central necrosis of granulomas, in addition to the epithelioid cell's apoptosis.22 The interaction of all these aspects and also the elements of the extracellular matrix9 are part of the final product, the granuloma with central caseous necrosis.23,24
Bacilli virulenceThe genome sequence of M. tuberculosis allowed to conduct studies about the role of unknown genes through functional genomics. The identification of genes present in hypervirulent strains of M. tuberculosis,25 but absent in pathogenic strains such as Mycobacterium bovis BCG, allowed a better understanding of the pathogenesis of TB.
Currently, the virulence of M. tuberculosis is defined by region of difference (RD1) genes, which are absent in BCG strains and are present in all M. tuberculosis virulent strains. Several studies have demonstrated that deletion of RD1 from M. tuberculosis results in attenuation of virulence, suggesting a dominant role in immune responses.26,27 Thus, deletion mutants of virulent M. tuberculosis strains for RD1 resemble BCG in their infectivity and attenuation.27
Furthermore, another group of genes have been associated with virulence of M. tuberculosis in animal model. It is suggested that the mammalian cell entry genes (mce), which encodes Mce protein, were originally identified in M. tuberculosis28 and have been associated with survival within macrophages and an increase in virulence.29,30 Disruption of the mce gene of BCG results in a mutant that exhibits reduced invasiveness epithelial cells, impairing their ability to survive and multiply inside phagocytic cells.31
Inhibition of macrophage apoptosis by M. tuberculosis strain H37Rv has been proposed as another virulence mechanism.32 This hypothesis is supported by studies that show that bacilli can cause necrosis instead of apoptosis in infected macrophages to avoid innate host defense. The macrophage cell death pathway by necrosis could be an exit mechanism for M. tuberculosis to facilitate the spread of infection and contribute to the formation of necrotic lesions in TB.33,34 Indeed, infection of macrophages with the attenuated M. tuberculosis causes apoptosis, which limits bacterial replications and promotes T-cell cross priming by antigen-presenting cells.35
Host hypersensitivityThere is a strong relationship among immunity and hypersensitivity in TB. T lymphocytes can produce distinct types of immune responses associated with different cytokine profiles and have opposing effects on resistance and susceptibility.14 The result of this balance will determine the degree of lesion in infected individuals and TB disease. Latent infection is associated with protective cellular immune responses whereas advanced disease is associated with immune suppression and exacerbated hypersensitivity reaction, causing tissue damage. Thus, T lymphocytes may also mediate harmful tissue-destroying hypersensitivity reactions that cause progression of disease.36
Natural and acquired resistanceMacrophages, dendritic cells and neutrophils principally mediate the natural host resistance to M. tuberculosis. The available data on the role of neutrophils are controversial. Neutrophils are the first cells to be mobilized and arrive within hours at the site of infection. They are not professional phagocytes such as macrophages, but contribute substantially to innate resistance to infection. Neutrophils can also form an extracellular fibril matrix known as NETs (neutrophil extracellular traps). These NETs were described recently.37 They are formed by activated neutrophils and consist of a DNA backbone with embedded antimicrobial peptides and enzymes. Thus, NETs represent a distinct innate defense mechanism to control and eliminate microbial infections.38 On the other hand, the poor ability of neutrophils to restrict mycobacterial growth compared to alveolar macrophages indicates that the prevalence of neutrophils in TB inflammation contributes to the development of a nonspecific cellular reaction, rather than protection of the host, and that neutrophils may play the role of a “Trojan horse” for M. tuberculosis.10,39 Indeed, interferon-inducible neutrophil-driven blood transcriptional signature has been described in patients with active TB, which correlates with lung radiographic disease severity and implicates neutrophils directly in the pathogenesis of active TB.40
Molecular studies show that the human genetic are responsible for natural resistance to M. tuberculosis. There is consensus in the literature that humans exposed to Mycobacterium millennially were evolutionarily selected, providing relatively more resistant cohorts. One example of this is the infection of Native Americans when Europeans first came to North America where the Apaches tribe was rapidly infected by acute and deadly TB.41 Also, indigenous Brazilians show a high TB rate, which can be attributable to an intrinsic susceptibility of these individuals.42
The resistance mechanism in TB involves a great number of cellular interactions (macrophage, T cell). Several different T-cell populations are required for the successful control of the pathogen. This dynamic interplay underlying protection is the reason for the long-term persistence of M. tuberculosis. From the cellular point of view the T lymphocytes are differentiated in the thymus. CD4 and CD8 T-cells activate macrophages against M. tuberculosis. These cells recognize specific parts of the bacillus and secrete cytokines, which activate macrophages.9,11–15,18 T cells also act, induced by components of the bacillus, producing defense mechanisms. By reacting with proteins, a state of hypersensitivity is created that can be measured by the tuberculin test. In this context, cytokines play an important role in the pathogenesis of TB. The cytokines are weighted molecules of about 8–14kDa whose production is a function of the components of M. tuberculosis. These cytokines attract macrophages that transform into epithelioid cells.8,9,14 IFN-γ, TNF-α and interleukin-(IL)-12 are the most fundamental cytokines, but there are currently about 40 classified cytokines and other chemokines that act as receptors involved in the cellular dynamic of granulomas.43
The events underlying the structural reorganization of immune cells to form a stable granuloma or progress to a pathologic lesion are dependent on other components. It is related with some components of the structure of M. tuberculosis, including some parts of its genetic material. These elements can mobilize, recruit, and amplify T cells reaction through the influence of cytokines.44 Glycolipids, components of the M. tuberculosis wall (sero-lipidic membrane), also involve the bacillus and DNA. They contain a 6kDa protein, designed as an ESAT-6, that mobilizes the T cell and activates the production of TNF-α and IFN-γ. Both of these activations lead the macrophages to destroy the Mycobacterium.19,45 Following the above events, as well as the arrival of bacilli in the tissues (for example, pulmonary alveoli), the formation of a granuloma with central caseous necrosis is completed.
Directed by T lymphocytes, modified monocytes aggregate and transform into epithelioid cells. With this fusion, giant multinucleated cells (Langerhans cells) are created. The dynamic previously described is also influenced by TNF-α.44 In addition to Langerhans cells, TNF-α also plays a role in the formation of caseous necrosis within the center of the granuloma.9,44 The anaerobic environment and the action of phagolysosomes within the granulomas inhibit the spread of bacilli. The hypersensitivity promotes the progression to necrosis, making the cells more sensitive to the components of the bacillus (for example, tuberculostearic acid). The final differentiation of the granuloma will depend on the interplay of all the factors cited in this paper. When there is high immunity and low hypersensitivity there will be a low level of necrosis and the granuloma will most likely scar under the action of miofibroblasts. If the hypersensitivity is dominant, the necrosis will spread and the lesion will grow with an increase in the proliferation of bacilli.46
In conclusion, although Rich's formula was proposed in the 1950s, it still holds some relevance to the distinct components within the granulomatous lesions. Even after incorporation of current knowledge about pathogenesis of M. tuberculosis, the balance between the characteristics of the bacillus and the host protective response is necessary to indicate the outcome of infection.
Conflict of interestThe authors have no conflict of interest to declare.
