


Latent HIV-1 is a major hurdle in obtaining HIV-1 sustained virological remission (SVR). Here we explored histone deacetylation inhibition property of nicotinamide (NAM; n=17) for the first time in comparison to a combination of methyltransferase inhibitors (MTIs; Chaetocin and BIX01294; n=25) to reactivate latent HIV ex vivo in CD8-depleted PBMCs from antiretroviral treated aviremic individuals.
ResultsNAM reactivated HIV-1 from 13/17 (76.4%) samples compared to 20/25 (80.0%) using MTIs with mean viral load (VLs) of 4.32 and 3.22 log10 RNA copies/mL, respectively (p=0.004). Mean purging time after NAM and MTIs stimulation was 5.1 and 6.75 days, respectively (p=0.73). Viral purging in autologous cultures exhibited blunted HIV recovery with fluctuating VLs followed by a complete viral extinction when expanded in allogenic system. Electron microscopy from five supernatants revealed anomalous viral particles, with lack of complete viral genomes when characterized by ultradeep sequencing through metagenomics approach (n=4).
ConclusionNAM alone was more potent HIV-1 activator than combination of MTIs, with potential of clinical use.
Even with effective antiretroviral therapy (ART), a cure for HIV remains elusive due to persistent viral replication in lymphoid tissues and latent resting CD4+ T lymphocytes.1
Being latent, HIV remains hidden from the immune cells and ART, and long lived resting memory CD4+ T lymphocytes are believed to represent the predominant reservoir of proviruses.2 Latency reversal agents (LRAs) have helped to interrupt the dormant state of HIV so that HIV expression can be induced, followed by clearance of that cell by viral cytopathic effects, immune clearance or antiretrovirals.3 Many distinct LRAs (Vorinostat, Romidopsin,4 Chaetocin,5 Panobinostat,6 BIX01294,7 Thalidomide8) have demonstrated the ability to inhibit chromatin condensation, restore suppressor gene expression and exert antitumor effects in many in vitro and in vivo studies. Hypoacetylation is one of the best characterized modifications where histone deacetylases (HDACs) cleave the acetyl group from amino groups of lysine residues in histone tails of nucleosomes. This results in condensed chromatin with less transcription factors accessibility, therefore silencing the gene. Cofactor dependence and sequence homology divide HDACs into two families and four different classes (IIV). The classic HDAC family (classes I, II, and IV) needs a zinc ion at the catalytic site to cause deacetylase activity while the Silent Information Regulator 2 (Sir2)-related protein (sirtuin) family (class III HDACs) requires nicotinamide adenine dinucleotide (NAD) as a cofactor for its catalytic activity and shows no sequence homology to classical zinc-dependent HDACs.9
There are many pathways that can convert NAM into nicotinamide mononucleotide (NMN) through the enzyme nicotinamide phosphoribosyltransferase (NMPRTase).10 NMN can then be directly converted into NAD by nicotinic acid/nicotinamide mononucleotide adenylyltransferase (NMNAT) which is active for NMN substrates.11 NAM released in the reaction inhibits SIRT1 through the so-called base exchange pathway, wherein rebinding of the reaction product to the enzyme accelerates the reverse reaction with an intermediate in the catalytic cycle instead of deacetylation reaction.12 Sirtuins are thus regulated by their cofactor NAD as well as their reaction product NAM that acts as their inhibitor.
The regulation of HIV-1 transcription is an interplay of both viral and host cellular components such as nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB).13 NFκB, being a critical regulator of HIV transcription, recruits the positive transcription elongation factor b (P-TEFb) to the viral 5’LTR, with the help of which only deacetylated form TAT protein binds to transactivation response element (TAR) that forms a stable RNA stem loop at the 5′ end of all viral transcripts.14 As the p65 subunit of NFκB and TAT are substrates of SIRT1,15 TAT binds to SIRT1 and is deacetylated. This binding might inhibit SIRT1’s further deacetylation activity and thus helps in setting p65 subunit of NFκB free to carry out its function, leading to T cell hyperactivation.16
Since NAM has been shown to affect SIRT1 activity,11,17 we used NAM to revert HIV latency in comparison to a combination of two MTIs, Chaetocin and BIX01294 that respectively inhibit SUV39 and G9a, with ability to revert HIV latency.5,7 We demonstrated that NAM alone is more efficient in breaking latency than a combination of MTIs, in comparing the time and viral loads of the samples ex vivo.
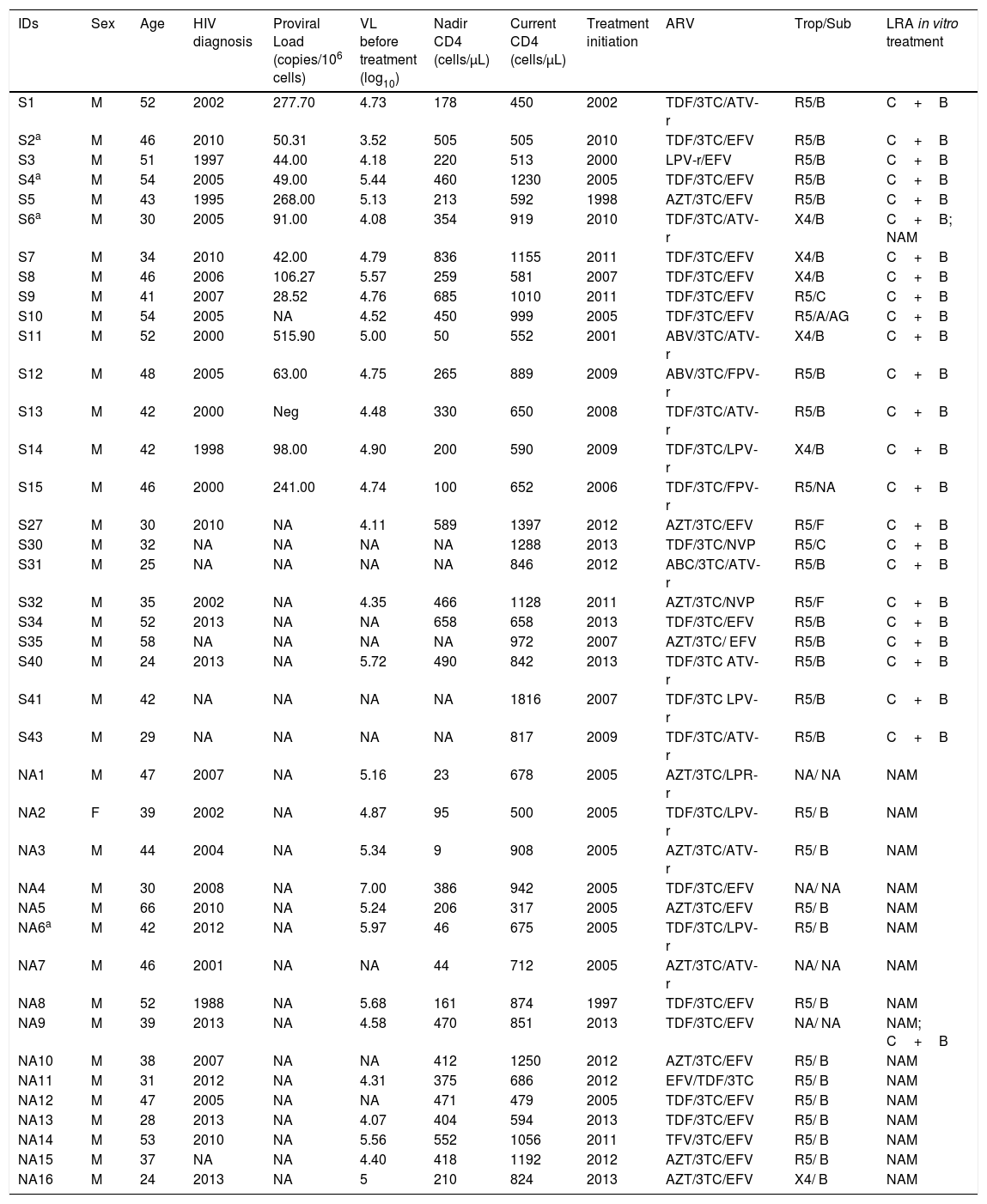
MethodsA total of 40 HIV-1 positive volunteers were recruited from the Aids Clinic of the Federal University of São Paulo, Brazil according to the following inclusion criteria: 18 years old or more, voluntarily signed Informed Consent Form (ICF), on ART with undetectable viral loads for at least three years, current CD4+T lymphocytes count greater than 350 cells/μL. This study and the ICF have been approved by the Ethical Review Board in Brazil (# number 226.613, CAAE 10757312.0.000.5505). Recruited individuals were also part of a clinical trial aiming the reduction of latent HIV proviral reservoir (www.clinicaltrials.gov; ID: NCT02961829). Characteristics of these patients are reported in Table 1.
Demographic, virologic and immunological characteristics of samples treated with MTIs and NAM ex vivo.
| IDs | Sex | Age | HIV diagnosis | Proviral Load (copies/106 cells) | VL before treatment (log10) | Nadir CD4 (cells/μL) | Current CD4 (cells/μL) | Treatment initiation | ARV | Trop/Sub | LRA in vitro treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | M | 52 | 2002 | 277.70 | 4.73 | 178 | 450 | 2002 | TDF/3TC/ATV-r | R5/B | C+B |
| S2a | M | 46 | 2010 | 50.31 | 3.52 | 505 | 505 | 2010 | TDF/3TC/EFV | R5/B | C+B |
| S3 | M | 51 | 1997 | 44.00 | 4.18 | 220 | 513 | 2000 | LPV-r/EFV | R5/B | C+B |
| S4a | M | 54 | 2005 | 49.00 | 5.44 | 460 | 1230 | 2005 | TDF/3TC/EFV | R5/B | C+B |
| S5 | M | 43 | 1995 | 268.00 | 5.13 | 213 | 592 | 1998 | AZT/3TC/EFV | R5/B | C+B |
| S6a | M | 30 | 2005 | 91.00 | 4.08 | 354 | 919 | 2010 | TDF/3TC/ATV-r | X4/B | C+B; NAM |
| S7 | M | 34 | 2010 | 42.00 | 4.79 | 836 | 1155 | 2011 | TDF/3TC/EFV | X4/B | C+B |
| S8 | M | 46 | 2006 | 106.27 | 5.57 | 259 | 581 | 2007 | TDF/3TC/EFV | X4/B | C+B |
| S9 | M | 41 | 2007 | 28.52 | 4.76 | 685 | 1010 | 2011 | TDF/3TC/EFV | R5/C | C+B |
| S10 | M | 54 | 2005 | NA | 4.52 | 450 | 999 | 2005 | TDF/3TC/EFV | R5/A/AG | C+B |
| S11 | M | 52 | 2000 | 515.90 | 5.00 | 50 | 552 | 2001 | ABV/3TC/ATV-r | X4/B | C+B |
| S12 | M | 48 | 2005 | 63.00 | 4.75 | 265 | 889 | 2009 | ABV/3TC/FPV-r | R5/B | C+B |
| S13 | M | 42 | 2000 | Neg | 4.48 | 330 | 650 | 2008 | TDF/3TC/ATV-r | R5/B | C+B |
| S14 | M | 42 | 1998 | 98.00 | 4.90 | 200 | 590 | 2009 | TDF/3TC/LPV-r | X4/B | C+B |
| S15 | M | 46 | 2000 | 241.00 | 4.74 | 100 | 652 | 2006 | TDF/3TC/FPV-r | R5/NA | C+B |
| S27 | M | 30 | 2010 | NA | 4.11 | 589 | 1397 | 2012 | AZT/3TC/EFV | R5/F | C+B |
| S30 | M | 32 | NA | NA | NA | NA | 1288 | 2013 | TDF/3TC/NVP | R5/C | C+B |
| S31 | M | 25 | NA | NA | NA | NA | 846 | 2012 | ABC/3TC/ATV-r | R5/B | C+B |
| S32 | M | 35 | 2002 | NA | 4.35 | 466 | 1128 | 2011 | AZT/3TC/NVP | R5/F | C+B |
| S34 | M | 52 | 2013 | NA | NA | 658 | 658 | 2013 | TDF/3TC/EFV | R5/B | C+B |
| S35 | M | 58 | NA | NA | NA | NA | 972 | 2007 | AZT/3TC/ EFV | R5/B | C+B |
| S40 | M | 24 | 2013 | NA | 5.72 | 490 | 842 | 2013 | TDF/3TC ATV-r | R5/B | C+B |
| S41 | M | 42 | NA | NA | NA | NA | 1816 | 2007 | TDF/3TC LPV-r | R5/B | C+B |
| S43 | M | 29 | NA | NA | NA | NA | 817 | 2009 | TDF/3TC/ATV-r | R5/B | C+B |
| NA1 | M | 47 | 2007 | NA | 5.16 | 23 | 678 | 2005 | AZT/3TC/LPR-r | NA/ NA | NAM |
| NA2 | F | 39 | 2002 | NA | 4.87 | 95 | 500 | 2005 | TDF/3TC/LPV-r | R5/ B | NAM |
| NA3 | M | 44 | 2004 | NA | 5.34 | 9 | 908 | 2005 | AZT/3TC/ATV-r | R5/ B | NAM |
| NA4 | M | 30 | 2008 | NA | 7.00 | 386 | 942 | 2005 | TDF/3TC/EFV | NA/ NA | NAM |
| NA5 | M | 66 | 2010 | NA | 5.24 | 206 | 317 | 2005 | AZT/3TC/EFV | R5/ B | NAM |
| NA6a | M | 42 | 2012 | NA | 5.97 | 46 | 675 | 2005 | TDF/3TC/LPV-r | R5/ B | NAM |
| NA7 | M | 46 | 2001 | NA | NA | 44 | 712 | 2005 | AZT/3TC/ATV-r | NA/ NA | NAM |
| NA8 | M | 52 | 1988 | NA | 5.68 | 161 | 874 | 1997 | TDF/3TC/EFV | R5/ B | NAM |
| NA9 | M | 39 | 2013 | NA | 4.58 | 470 | 851 | 2013 | TDF/3TC/EFV | NA/ NA | NAM; C+B |
| NA10 | M | 38 | 2007 | NA | NA | 412 | 1250 | 2012 | AZT/3TC/EFV | R5/ B | NAM |
| NA11 | M | 31 | 2012 | NA | 4.31 | 375 | 686 | 2012 | EFV/TDF/3TC | R5/ B | NAM |
| NA12 | M | 47 | 2005 | NA | NA | 471 | 479 | 2005 | TDF/3TC/EFV | R5/ B | NAM |
| NA13 | M | 28 | 2013 | NA | 4.07 | 404 | 594 | 2013 | TDF/3TC/EFV | R5/ B | NAM |
| NA14 | M | 53 | 2010 | NA | 5.56 | 552 | 1056 | 2011 | TFV/3TC/EFV | R5/ B | NAM |
| NA15 | M | 37 | NA | NA | 4.40 | 418 | 1192 | 2012 | AZT/3TC/EFV | R5/ B | NAM |
| NA16 | M | 24 | 2013 | NA | 5 | 210 | 824 | 2013 | AZT/3TC/EFV | X4/ B | NAM |
C+B: Chaetocin+BIX01294; NAM: Nicotinamide; ID: individuals identification; NA: Not available; VL: viral load (copies/mL); ARV: antiretrovirals; TDF: tenofovir; AZT: zidovudine; ABV: abacavir; 3TC: lamivudine; EFV: efavirenz; NVP: nevirapine; ATV-r: ritonavir boosted atazanavir; LPV-r: ritonavir boosted lopinavir; FPV-r: ritonavir boosted Fosamprenavir; Trop: Co-receptor tropism; X4: non-R5 tropic HIV; R5: R5 tropic virus; Sub: Viral subtype.
PBMCs from HIV-infected individuals and healthy donors were isolated by Ficoll-Hypaque gradient (GE Healthcare, Uppsala, Sweden) from 100mL of whole blood. To avoid cytotoxic effects in cultures, CD8 T cells were depleted by EasySep Human CD8 Positive Selection kit (STEMCELL TECHNOLOGIES) and were cryopreserved in fetal bovine serum (FBS; Gibco) with 10% DMSO.
CD8-depleted PBMCs using the EasySep Human CD8 Positive Selection kit II (STEMCELL TM TECHNOLOGIES, Vancouver, Canada) were treated at a density of 107 cells with either 10mM NAM18 or a combination of 90nM Chaetocin19 and 10μMBIX-012947 for 48h and cultivated in RPMI medium (supplemented with 10% FBS and IL-2 20 IU/mL). Cell viability was assessed by cell counting after staining with trypan blue, and the cells were also closely monitored under the microscope. If the number of cells was on increase, it was assumed that the cells were proliferating and there was no drug associated cytotoxic effects. No drug associated cytotoxicity was observed after the treatment. Cells were always clumped together (healthy cells) rather than isolated cell suspension, and the overall viability was always above 70%.
After 48h, cells were washed to remove excess PHA and LRAs and were cultivated in RPMI. Cultures were fed weekly with 107 CD8-depleted autologous PBMCs, previously activated with PHA and treated with LRAs for 48h. We assumed that extracellular virions would accumulate over the course of several days; therefore, the supernatants were withdrawn every 3–4 days, while replenishing the cultures with fresh medium, for monitoring viral purging by the quantification of viral RNA with qPCR (Abbott Real Time HIV-1 assay, Abbott, Chicago, IL, USA). Viability was always checked before starting the culture using Neubauer chamber with trypan blue stain and was 90–95%.
We separated 2×107 cells from samples S1-S5 (samples C.S1 - C.S4; Fig. 1A) and NA1-NA4 (C.NA1-C.NA4; Fig. 1B) and used as controls without LRAs. Due to limited numbers of autologous cells, the controls were kept over a period of 10 days only, seeded twice with 107 PHA activated autologous cells. The reagents Chaetocin (C9492), BIX-01294 (B9311) and Nicotinamide (N3376) were purchased from Sigma Aldrich (St. Louis, USA).
 Graph showing the Log10 viral load (copies/mL) of the culture supernatant treated with MTIs, (B) Graph showing the Log10 viral load (copies/mL) of the culture supernatant treated with NAM, (C) Comparison of NAM+MTIs to break latency with respect to time. (D) Comparison of NAM+MTIs with respect to viral load.")
Ex vivo treatment with MTIs and NAM and their comparison.
(A) Graph showing the Log10 viral load (copies/mL) of the culture supernatant treated with MTIs, (B) Graph showing the Log10 viral load (copies/mL) of the culture supernatant treated with NAM, (C) Comparison of NAM+MTIs to break latency with respect to time. (D) Comparison of NAM+MTIs with respect to viral load.
Depending upon the availability of PBMCs, buffy coat or culture supernatants, total DNA and RNA were extracted using Qiagen DNA Blood Mini Kit (Qiagen, Hilden, Germany) and Qiagen Viral RNA Mini Kit (Qiagen, Hilden, Germany), respectively, following the manufacturer´s instructions.
HIV proviral loads were measured by qPCR using the HIV-1 LTR sequence as the target and the human CCR5 gene as the normalizer. We extrapolated copy numbers from standard curves generated with plasmids harboring the CCR5 and the HIV sequence, as previously described.20 The reaction mixture consisted of 20μL of Gene expression master mix (Life Technologies, Carlsbad, CA, and USA Applied Biosystems, Coutabouef, France) 2X, 0.75μM of each oligonucleotide, 0.3μM of each probe, 5μL of sample, and DNase RNase-free water with total volume of 20μL. Sequences of primers and probes and cycling conditions have been previously described.20
Viral tropism was inferred either by proviral DNA or viral RNA extracted from culture supernatants as previously described.21,22 The generated DNA sequences were aligned and edited manually for the codon alignment using BioEdit Version 7.2.5. The Geno2pheno[correceptor] algorithm (available at http://www.coreceptor.geno2pheno.org) with an FPR cutoff of 10% was used to infer co-receptor tropism.
Metagenomics methodology was implied to de novo characterize supernatant samples containing purged HIV-1 particles following in vitro treatment with LRAs. RNA from supernatant was extracted using QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacture’s instruction. Extracted RNA was followed by reverse transcription into cDNA using random primers and SuperScript III-Reverse transcriptase (Invitrogen) and the pre-amplification step was performed to enrich the viral nucleic acids as previously described.23 Libraries were prepared using Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) and sequenced using Illumina MiSeq platform.
Sequencing reads with Phred quality score over 30% (i.e., a base call accuracy of 99.9%) were aligned using Tanden (Tool for Analysis of Diversity in Viral Populations) software that performs the mapping and indexing of each short read, being utilized in data banks with 1258 reference sequences, including representatives of group M of HIV with all subtypes and recombinants, HIV group O and some SIVcpz sequences. The aligned sequences were validated according to their E-values and the distance from the reference sequence.
p24 was quantified in culture supernatants by ELISA ZeptoMetrix kit (ZeptoMetrix Corporation, Buffalo, New York), according to manufacturer’s instructions.
Morphology of HIV particles was analyzed by TEM in fresh culture supernatants. TEM was performed as previously described with modifications.24 Briefly, formvar-carbon-coated 300 mesh copper grids were treated with 30μL of Alcian Blue solution in 1% acetic acid for 10min. Then, the grids were rinsed using three drops of sterile distilled water. After removal, with filter paper, grids were placed onto 30μL of culture supernatants for 10min. RPMI was used as negative control. Staining was carried out by successively transferring the grids to three drops of 2% potassium phosphotungstate (ph 7.2), dried with filter paper, and keeping at least 24h at room temperature before the analysis mission electron microscopy. Grids were observed under a JEOL TEM (model JEM1011; JEOL/Massachusetts/USA) operating at 80kV. Images were recorded with a charged-couple device camera (model 785 ES1000W, Gatan, USA) and the Gatan program version 1.6.
All statistical analyses were conducted using the R Statistical Language and Computing System, version 3.3.2. Kruskal-Wallis rank sum non-parametric chi-squared test were performed using Desc function of the DescTools package. In all cases a p-value of ≤ 0.05 was considered statistically significant. All the Graphs were developed using GraphPad Prism (v6.0).
ResultsNAM is more potent activator of HIV in ex vivo primary cultures than a combination of MTIs (Chaetocin and BIX01294)To assess LRAs for their ability to bring HIV out of latency, we used previously described MTIs and a novel SIRT1 inhibitor, NAM, in ex vivo cultures of CD8-depleted PBMCs from HIV-1 infected, ART treated patients who had undetectable viral loads over a period of at least three years. HIV latency reversal was detected using a commercial viral load assay in the supernatants from cultures by qPCR using the Automated 2000 System (Abbott, IL, USA), which has a dynamic range of detection from 40 to 1×107 copies of the HIV-1 RNA/mL. In terms of virus reactivation, both LRAs schemes displayed varying levels of potency in inducing HIV expression, although a wide standard deviation was observed in the magnitude of viral purging for both LRAs schemes (Fig. 1D).
Among samples treated with MTIs, 20/25 samples (80%) showed viral purging with a mean duration of 6.75 days (range: 3–29; Fig. 1A) and among the samples treated with NAM (n=17), thirteen (76.4%) showed positivity of the cultures as inferred by the viral load result, with a mean time of 5.1 days (range; 3–17; Fig. 1B). No HIV-1 recovery was detected in control samples, which were not subjected to the action of the LRAs used in the experiment.
We then compared the purging potential of NAM and MTIs by considering time duration that LRAs took to break latency and their resulting viral loads. The proportion of positive samples treated with NAM was superior to the proportion of MTIs in the earliest category of purging – less than three days (52.8% for NAM against 40% for the MTI). Thereafter, the two types of treatment were roughly equivalent in the proportion of samples that demonstrated purging. Therefore, considering time across all samples tested for each LRA to break latency, no significant statistical difference was observed between the two groups (Chi-square test; n=42, p=0.7323 (2-tailed); Fig. 1C).
We compared the viral loads of supernatants at first positive collection. In this case, NAM showed higher mean log10 viral load (4.32 copies/ml) than MTIs (3.22 copies/ml) and this difference was found to be significant (Chi-square test, p=0.004551; Fig. 1D). These findings suggest that activation of HIV transcription with NAM is more potent than MTIs.
LRAs manifest variable ex vivo HIV-1 activation in longitudinal samples from the same individualIn order to determine whether HIV-1 reactivation by these LRAs was consistent in samples collected longitudinally from the same patient, four samples were selected that showed no HIV-activation in initial attempts. RNA levels were measured from these samples treated with LRAs from the same individuals collected several months apart. Two of the tested samples (S2 and S4) showed HIV-1 activation at third attempt after the treatment with MTIs. While the other two samples (S6 and NA9) remained negative despite several attempts of the treatment with MTIs or NAM alone or even in combination. These findings showed that there are inter-patient variations in ex vivo HIV activation by MTIs or NAM.
Viral purging is independent of proviral load and viral tropismWe were able to quantify proviral DNA in 14 out of 25 individuals treated with MTIs in vitro with a median of 140.2 copies/106 PBMCs (range: 28.5–515.9). In 3/5 of negative cultures one sample was also negative for proviral load (Table 1). From both LRAs treatment, approximately 80% of the individuals showed R5 tropism (Table 1). However, no correlation was found between proviral load/viral tropism and purging (Supplementary Fig. S1).
Expansion of the virus in allogenic systemTreatment with both MTIs and NAM exhibited a common feature in terms of viral purging: blunted HIV recovery with fluctuating viral loads that either increased over time or declined, despite cultures being continually fed with new CD8-depleted and PHA-activated/LRAs treated PBMCs (Fig. 1A and B). Seven samples treated with MTIs and five treated with NAM showed an increase in viral load over a course of 29 and 18 days, respectively. On the other hand, seven samples of MTIs group and four from NAM showed decrease in viral loads.
Sample (S31) treated with MTIs showed abnormal behavior in the culture. It turned positive at day 10 (third collection) but returned to negative in the next two subsequent collections although the culture was fed with new round of MTIs treated CD8-depleted autologous PBMCs and the medium was being replenished every third or fourth day. It turned positive again at day 21 (sixth collection) when it was fed for the third time with a new round of cells. At the seventh collection, the viral load was undetectable again showing extinction of the virus according to viral load assay. This sample turned positive again at ninth collection after a fourth round of feeding with autologous cells, presenting a viral load of 10,014 copies/ml (Fig. 1A).
We hypothesized that this fluctuation of viral loads could be related to some sort of host restriction interfering with the viral replication. To resolve this, we selected nine samples and expanded these viruses in allogenic system and observed the same behavior of the viruses, showing declining of viral load and ultimate extinction (Supplementary Table S1).
Cultures were repeated with a lower multiplicity of infection (MOI) in order to avoid the impact of defective interfering particles. Again, after an initial HIV recovery, cultures evolved to a rapid HIV extinction (data not shown). This excluded the possibility of host specific restrictions but raised suspicion that purged viruses among these ART suppressed individuals might be defective.
p24 QuantificationIn order to check the specificity of HIV-viral loads in quantifying alleged defective strains, 18 samples were selected with low, intermediate, and high viral load to quantify p24 antigen. p24 could only be quantified in those samples that had intermediate and/or high viral loads with proportional results between the two systems (Supplementary Table S2).
Viral extinction correlates with defective virions observed by EMAfter observing anomalous behavior of the virus in autologous as well as allogenic system, we visually inspected the virus by EM (Fig. 2E and F). We randomly selected five supernatant samples. EM revealed abnormal features in all inspected viral particles, such as smaller size (30−90nm in diameter), distinct electron dense central cores could not be seen and fully double ring particles were very infrequent. The projections on the viral envelope could not be clearly discerned, in some cases particles were doughnut shaped double ringed structure with an electron lucent center.25
 and NAM (patients NA5 and NA8, panels B and D) showing uneven coverage of HIV genome. Panels E and F shows the electronic microscopy result performed in the supernatant of samples S2 and NA8 respectively showing anomalous HIV particles.")
Characterization of the purged viruses using Next Generation sequencing with Metagenomics Methodology for de novo HIV characterization on supernatant samples containing purged HIV-1 particles of 4 selected samples following in vitro treatment with the MTIs association (patients S7 and S2, panels A and C) and NAM (patients NA5 and NA8, panels B and D) showing uneven coverage of HIV genome. Panels E and F shows the electronic microscopy result performed in the supernatant of samples S2 and NA8 respectively showing anomalous HIV particles.
The abnormal viral propagation in ex vivo cultures and compromised morphology seen by EM was suggestive of massive presence of defective particles post LRA stimulation. To confirm this, metagenomics UDS was performed. According to this approach, an equal number of reads throughout the HIV-1 genome was expected since random primers were used. However, we were not able to recover full-length genome data from the cell culture supernatants by UDS methodology and analysis. The number of reads was completely uneven which suggests that HIV-1 genome from the supernatant contains major deletions causing the particles to be defective, as previously described.26 Although the coverage was not optimal but in two samples, S7 and NA5, treated in vitro with MTIs and NAM, respectively, we were able to recover partial to complete genomes (Fig. 2A and B). NA5 showed missing patches of gag and nef in its genome. While the sequencing data from NA8 showed three stretches of envelope gene encompassing position 6920-8271bp (Fig. 2D) and S2 showed partial complete genome encompassing 2389–8078 bp (Fig. 2C). This lack of complete genome in these two samples is in agreement with anomalous viral particles seen by EM (Fig. 2E and F) and extinction of the viruses observed in allogenic system (Supplementary Table S1).
Altogether, the whole sequencing data suggests that viral particles purged after NAM and MTIs stimulation from patients had packaged incomplete HIV-genome in them. We understand that this virus is not able to make a complete next round of infection (Additional file 1: Table S1).
DiscussionThis is the first demonstration in HIV-infected ex vivo primary cell cultures that NAM, a Vitamin B, causes purging of dormant provirus with short-term treatment and notably high virus production. Considering reactivation in ex vivo assays with different LRAs like Chaetocin, BIX01294, Romidopsin, Panobinostat, and Bryostatin, we observed significantly higher virus production in the cultures treated with NAM. However, direct comparison with Panobinostat, Romidopsin, and Bryostatin has not been performed in this study.
We used two already reported MTIs, Chaetocin and BIX01294,19 and NAM to compare combination of two LRAs (MTIs) versus one NAM. A significant difference was observed in culture viral loads post-treatment (Fig. 1D) and purging of virus in terms of the time required for virus to emerge in the culture (Fig. 1C), although the wide standard deviation in viral loads after use of LRAs reinforces the unpredictable nature of the magnitude of viral purging. Cultures that stayed negative post MTIs treatment were kept for a mean time of 20 days (14–30 days) and in NAM for a mean time of 10 days only (7–17 days). It is conceivable that if cultures treated with NAM were also kept and fed for a longer period as were kept for MTIs, more efficient purging of the viruses from NAM treated samples would occur. Unfortunately, these cultures could not be maintained for longer periods due to unavailability of autologous cells for continuous feeding.
To find a correlation between viral purging and the proviral compartment, we were able to quantify proviral DNA in 14 available samples exposed ex vivo to MTIs. Sample (S13) remained negative for proviral load as well as in ex vivo culture. Therefore, we hypothesized that the HIV proviral compartment of this individual might be very low, preventing an efficient in vitro purging using MTIs. However, in the remaining 13 samples, no correlation was found between proviral loads and efficacy of LRA, time to virus purging and viral load at purging.
In vitro stimulation of T-lymphocytes by cytokines, lymphokines and mitogens contributes to their activation and proliferation by activating transcription factors such as NFκB. The HIV-1 promotor contains two tandem NFκB binding sites (κ response elements). Besides HIV-1, the IL-2 promotor also contains binding sites for NFκB.27 Thus, NFκB binding to its response elements can lead to the activation of respective promotors. Also, the p65 subunit of NFκB helps in activation of the HIV promotor by recruiting PTEFb (CDK9, Cyclin T1)28 which phosphorylates C-terminal domain (CTD) of RNA polymerase II.16 Thus, once a T cell is stimulated, it allows resting CD4+ T cells to undergo a burst of cellular proliferation and differentiation, giving rise to effector cells and thus breaking latency.
Although direct molecular mechanism using specific inhibitor/antibody of SIRT in combination with NAM has not been performed here, literature suggests that NAM can act as an LRA in several ways. (i) NAM is a deacytylase inhibitor because it blocks SIRT1, a class III HDAC, and thus inhibits deacetylation of histones and the consequent formation of facultative heterochromatin [reviewed in Ref. 11]. (ii) Alternatively, NAM may be directly salvaged back to NAD during biosynthesis and can act as an indirect activator of SIRT1.29 Its activation can break latency in two different ways. First, it can deacetylate various substrates, like the viral TAT protein30 and the p65 subunit of NFκB. TAT binds to SIRT1 and blocks deacetylation of the p65 subunit.17 This makes p65 subunit remain free and it can recruit PTEFb which phosphorylates CTD of RNAPII causing promotor activation and latency break. The deacetylated form of TAT, which is the only form able to engage in the formation of the TAT/TAR/P-TEFb complex, is thus available to the cell when SIRT1 is catalytically active in the presence of NAD. Secondly, SUV39, a methyltransferase, is another substrate of SIRT131 that favors latency by adding a methyl group to the histone proteins. Therefore, active SIRT1 can also deacetylate SUV39 thus avoiding chromatin condensation resulting in latency break.
Over 90% of proviruses in the peripheral blood are considered to be defective due to lethal genetic mutations that include G-to-A hypermutations and small insertions/deletions that interrupt ORFs, or even large deletions.32 It has been recently demonstrated that during long-term ART, the proportion of replication competent proviral compartment decreases over time as compared to defective HIV33 which suggests that replication competent provirus are coming out of latency over time34 leading to cell death by cytopathic effects and/or immune clearance. The participants of this study are under long-term ART with a mean duration of nine years (range: 3–20) conceivably harboring massive presence of defective provirus, confirmed by propagation of virus in autologous (Fig. 1A and B) as well as in allogenic system (Supplementary Table S1) and viral particles observed by electron microscopy which revealed anomalous HIV particles (Fig. 2E and F). Small sized particles seen by EM might have been purged from the cells harboring defective small proviral genome. We recognize that the absence of a positive control sample, such as a laboratory-adapted virus strain using a standard packaging cell line to compare with the patient-derived HIV, may preclude the conclusions whether some external factors could be altering the virus particles morphology analyzed by electronic microscopy or the “de novo” genomic sequencing results. However, several reports have demonstrated that the defective HIV obtained from individuals under long-term ART may present as small as 2kb proviral DNA.26,35 This is consistent with the finding of partial to complete genomes in our samples by UDS. From a virologic point of view, it is non-infectious material of no consequence. However, from an immunologic standpoint, it is the by-product of a viral mechanism to stay one-step ahead of the cellular and humoral responses.
The potential of these MTIs and NAM are evident by virus production shortly after stimulation of the samples with undetectable viral loads. However, we recognize some weaknesses of this study. We have not been able to use purified cell populations to determine the source of viral emergence. In addition, the limited number of cells from the same HIV donor precluded comparison by pairing the samples for both purging strategies to better determine the efficacy of each LRA separately or in synergy. Among the negative samples, only NA9 and S6 could be tested for both LRAs, treated separately and in combination to see any synergistic effect, if existed. However, no viral purging was observed. Individual NA9, a 39-year old male, was treated early in his HIV infection, with a recent seroconversion (HIV negative EIA six months before diagnosis and treatment initiation) and a CD4+ T lymphocytes count of 851 cells/mm3. This individual presented only one pre-treatment positive viral load of 37,660 copies/mL and was exposed to Tenofovir, 3TC and Efavirenz for three years. By the time the samples were drawn for HIV culture, a rapid HIV EIA test was repeatedly negative in the plasma sample (TR DPP® HIV1/2, Bio-Manguinhos, Rio de Janeiro, Brazil), although a regular 4th generation EIA test and Western blot were positive, which suggest a low level of HIV antibodies. Furthermore, nested PCR for the V3 region of the envelope gp120, envelope gp41, protease and reverse transcriptase of pol gene, and gag P17 were all negative for this patient. This individual presented a wild type CCR5 allele (data not shown). S6 was nested PCR positive for the V3 region of the gp120, with a genotype compatible with a non-R5 strain, and was heterozygous for CCR5 (CCR5Δ32/wild type). Considering that viral purging is interplay between (i) host transcription factors, (ii) LRAs, and (iii) viral protein (TAT), it is not clear whether culture negativity was related to a specific host restriction, overwhelming presence of defective viruses, viral integration site, epigenetic state, or lack of proper interaction between the drugs and latent cells. In addition, it is important to realize that the fate of cells expressing pharmacologically reactivated proviruses is not fully understood i.e., reactivation of latent HIV with NAM or other LRAs would lead to virus-induced cell death by direct or bystander mechanisms.
Our ex vivo results essentially brought NAM use as anti-latency drug. However, further studies are needed to determine its dose dependent effects on viral load, immune function and clinical outcomes in virally suppressed individuals. It should be noted that SIRT1 has a complex biology given the multiplicity of activities attributed to its sub-cellular localization and multitude of substrates. It has gained considerable interest by the pharmaceutical industry as a prospective drug target for the treatment of metabolic disease, cancer, inflammation, and/or neurodegeneration.36 Owing to SIRT1 complexity, appropriate animal models will also be important to understand NAM/SIRT1 biology in context of HIV infection to break latency. More potent and selective inhibitors of SIRT1 will need to be tested in vivo to unequivocally answer this question. Of note, NAM has been used in clinical medicine for a number of distinct purposes with a very safe profile.37
In conclusion, our observations provide the first evidence that NAM has a latency reversal activity with a potency and efficacy conceivably superior to the combination of two MTIs. NAM acts as a double edge sword as it can break HIV-1 latency by directly inhibiting SIRT1 or indirectly via NAD biosynthesis as an intermediate compound and activating SIRT-1. Thus, early purging induced by NAM with significantly high viral load could be due to more than one mechanism being stimulated simultaneously. Decrease in NAD level has been reported in HIV infection18 and since NAM has a central role in cellular metabolism, it is plausible that NAM supplementation can restore NAD levels and might also help in regulating the redox environment among HIV infected individuals. In addition, our data suggests that cells harboring defective proviral pool can produce defective viral particles, once they are stimulated and may be responsible for seropositivity, persistent immune activation and residual viremia among individuals with suppressed viremia. New latency reversal strategies must be designed aimed at eliminating those cells harboring this defective proviral pool to obtain HIV SVR.
Authors’ contributionsSS, TMO. SVK, LMJ, MCS and RSD designed the experiments. SS, MSA, LBG, BTL, GN and JG performed the experiments. SS, TMO, SVK, MCS, LMJ and RSD analyzed the data. SS and GN performed electron microscopy. JH did the statistical analysis. JPLZ performed NGS analysis. SS, MSA, LBG, JPLZ, JH, BTL, GN, LMJ and RSD prepared the manuscript. All authors reviewed the manuscript.
Competing financial interestsThe authors have declared that no competing interests exist.
Ethics approval and consent to participateThis study and the ICF have been approved by the Ethical Review Board in Brazil under the number 226.613, CAAE 10757312.0.000.5505. Informed written consent was obtained from all patients.
Consent for publicationAll authors have read and approved the manuscript for publication.
FundingThis work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 2013/11323-5) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 454700/2014-8). SS, MAS, LBG and JPZ received PhD fellowship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazilian Ministry of Education. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materialAll the sequence data generated in this study have been submitted to NCBI Genbank Database. Accession numbers (pending).
We are grateful to all HIV-infected patients who participated in the study by donating their blood.
The following is Supplementary data to this article:
