







Cardiac hypertrophy and dysfunction are a significant complication of chronic Chagas disease, with heart failure, stroke, and sudden death related to disease progression. Thus, understanding the signaling pathways involved in the chagasic cardiac hypertrophy may provide potential targets for pharmacological therapy. Herein, we investigated the implication of focal adhesion kinase (FAK) signaling pathway in triggering hypertrophic phenotype during acute and chronic T. cruzi infection. C57BL/6 mice infected with T. cruzi (Brazil strain) were evaluated for electrocardiographic (ECG) changes, plasma levels of endothelin-1 (ET-1) and activation of signaling pathways involved in cardiac hypertrophy, including FAK and ERK1/2, as well as expression of hypertrophy marker and components of the extracellular matrix in the different stages of T. cruzi infection (60–210 dpi). Heart dysfunction, evidenced by prolonged PR interval and decrease in heart rates in ECG tracing, was associated with high plasma ET-1 level, extracellular matrix remodeling and FAK signaling activation. Upregulation of both FAK tyrosine 397 (FAK-Y397) and serine 910 (FAK-S910) residues phosphorylation as well as ERK1/2 activation, lead to an enhancement of atrial natriuretic peptide gene expression in chronic infection. Our findings highlight FAK-ERK1/2 signaling as a regulator of cardiac hypertrophy in Trypanosoma cruzi infection. Both mechanical stress, induced by cardiac extracellular matrix (ECM) augment and cardiac overload, and ET-1 stimuli orchestrated FAK signaling activation with subsequent activation of the fetal cardiac gene program in the chronic phase of infection, highlighting FAK as an attractive target for Chagas disease therapy.
Chagas disease (CD), a neglected tropical disease caused by Trypanosoma cruzi, is considered an important public health problem responsible for high rates of morbidity and mortality in Latin America.1 Epidemiological surveillance has estimated that 8 million people are chronically infected worldwide, with more than 10,000 deaths per year.2 Acute infection is mostly asymptomatic, whereas chronic infection may remain without signs (indeterminate form), in 60–70% of infected people, or progress to distinct clinical symptoms as neurological, digestive (megacolon and megaesophagus) and heart manifestations.3–5 Cardiomyopathy is a relevant manifestation of CD, affecting 30% of infected individuals, and endemic countries such as Brazil (1.2 million infected people) and Argentina (1.5 million infected people) concentrate 42% of all Chagasic cardiopathy cases.6 Cardiac damage is progressive and classified into different stages (A, B, C and D) according to the severity of cardiac dysfunction.7 Chronically infected individuals with an unaltered electrocardiogram (ECG) are considered in the indeterminate phase of CD (stage A) while disease progression leads to ECG abnormalities (stage B), including arrhythmias or conduction disorders (B1) and decreased left ventricular (LV) ejection fraction (B2), and also signs of heart failure (stage C and D).7 Sudden cardiac death due to CD cardiomyopathy reaches annual mortality rates between 0.2 and 19.2%,8 highlighting the urgency of managing this silent disease in public policies.
Myocardial hypertrophy, an important pathological outcome of chronic chagasic cardiomyopathy (CCC), is an adaptative response to cardiac hemodynamic overload triggered by multifactorial processes, including cardiac injury due to severe myocarditis and fibrosis.9 Multiple signaling pathways, including NF-kB, mediated by Toll-Like receptor 2 (TLR2) that modulates pro-inflammatory cytokines (IL-1β),10,11 phosphatidylinositide 3-kinases (PI3K)/AKT/Nitric oxide (NO) elicited by increased tumor necrose factor-alpha (TNF-α) and/or NF-kB12 and also mitogen-activated protein kinase (MAPK) through extracellular signal-regulated protein kinases 1 and 2 (ERK1/2)13–15 or Ca2+/Calcineurin/nuclear factor of activated T cells (NFAT)-dependent mechanism16 mediated by endothelin-1 (ET-1), have been implicated in the upstream signaling of cardiac hypertrophic effectors induced by T. cruzi, resulting in the activation of hypertrophic-responsive genes that regulate cardiac growth.17
ET-1, a vasoactive peptide that acts through G-protein coupled receptors (GPCRs), endothelin receptor A (ETAR) and B (ETBR), is overexpressed in both acute and chronic phases of CD showing elevated plasma levels and high cardiac tissue concentration.18,19 ET-1, that acts in an autocrine and paracrine manner, potentiates the pro-inflammatory process by up-regulating cellular adhesion molecules (ICAM-1, VCAM-1, and e-selectin), favoring immune system cell traffic and thereby exacerbating myocardial cell damage.17 Persistence of the parasite in the chronic phase of the disease modulates the inflammatory response that sustains high levels of ET-1, capable of inducing cardiac hypertrophy and fibrosis. A remarkable feature is that ET-1 also modulates hypertrophic phenotype in dilated idiopathic cardiomyopathy (DCM) triggered by focal adhesion kinase (FAK).20 However, the participation of the FAK signaling pathway in chagasic cardiac hypertrophy has yet not been evaluated.
FAK, a non-receptor protein tyrosine kinase essential for cardiac physiology, has emerged as a protagonist molecule of hypertrophy and heart failure.21,22 FAK signaling regulates mechanotransduction and sustains cardiac hemodynamics through integrin signaling pathway23 but may also be activated through transmembrane receptors as heparan sulfate proteoglycans (HSPG)24 and GPCRs,25 the latter has been suggested as an activator of the hypertrophic response mediated by ET-1.26 FAK is a multidomain scaffold protein composed of a band 4.1, ezrin, radixin, moesin (FERM) domain, a central kinase domain and a focal adhesion targeting (FAT) domain. The autophosphorylation of FAK tyrosine residue 397 (Y397) triggers the recruitment and activation of Src-kinase family amplifying kinase activity through the phosphorylation of other tyrosine residues (Y576/577, Y861 and Y925), regulating biological events as adhesion, migration, proliferation, and survival.27,28 Additionally, FAK serine residues phosphorylation (S722, S843, S846, and S910) modulates hypertrophic response29 and, interestingly, ET-1 is a key inducer of FAK-S910 phosphorylation promoting sarcomere reorganization and cardiac hypertrophy,20 thus raising questions about the role of FAK signaling cascade in triggering chagasic cardiac hypertrophy.
In this study, we evaluated whether FAK signaling may act as a regulator of cardiac hypertrophy in Chagas cardiomyopathy. FAK activation was assessed by FAK-Y397 and FAK-S910 phosphorylation and its downstream effector in both acute and chronic phases of the murine model of T. cruzi infection.
Materials and methodsT. cruzi experimental infectionC57BL/6 lineage male mice (15–20g), obtained from the Institute of Science and Technology in Biomodels (ICTB, Fiocruz), were housed at a maximum of five animals per cage, kept in a specific-pathogen-free (SPF) room at 20–22°C under a 12/12h light/dark cycle, 50–60% humidity and provided sterilized water and chow ad libitum. All experimental animal procedures were performed following the license (L–015/2017) approved by the Ethics Committee for Animal Use of the Oswaldo Cruz Institute (CEUA/IOC) and also following Brazilian Law 11.794/2008 and regulations of the National Council of Animal Experimentation Control (CONCEA). In order to preserve animal welfare, a daily observation was supervised by a PhD veterinary doctor aiming to avoid animal suffering and pain. Animals were humanely euthanized whenever early endpoints of animal suffering became apparent such as motor disturbance, exploratory activity and/or moribund condition.
A total of 42 mice was divided into two groups: noninfected (12) and T. cruzi-infected (30) animals. Mice were intraperitoneally (i.p.) infected with trypomastigotes forms of T. cruzi Brazil strain (105 parasites/animal), while noninfected animals were inoculated with sterile phosphate-buffered saline (PBS). Parasitemia was evaluated by Pizzi-Brener method.30 Briefly, fresh blood samples (5μL) were collected by the incision of the tail vein and examined by light microscopy. The circulating parasite load was monitored daily from 5th day of infection until 50 days post-infection (dpi). Acute infection (30–60 dpi) was determined by positive parasitemia and tissue parasitism. The surviving animals were monitored during the course of the chronic infection (90–210 dpi).31 The heart was harvested from both noninfected and T. cruzi-infected mice from 60 to 210 days. After longitudinally sectioned, half of the cardiac tissue was cryopreserved with Tissue-Tek for immunohistochemical analysis and the remaining tissue was fragmented and processed for protein and RNA extraction. Two independent experiments were performed.
Electrocardiographic analysisElectrocardiographic parameters were evaluated in non-sedated mice by using transducers carefully placed under the skin, in accordance with a chosen preferential derivation (DII). Traces were recorded with a digital system (Power Lab 2/20) connected to a bio-amplifier at 2mV for 1s (PanLab Instruments). The filters were standardized between 0.1 and 100Hz, and the traces were analyzed by using Scope software for Windows V3.6.10 (PanLab Instruments). We measured cardiac frequency (beats per minute, bpm) and duration of the PR, QRS, and QT intervals in milliseconds (ms) through (i) P wave: atrium chambers, (ii) PR interval: atrioventricular electric conduction, (iii) QRS interval: ventricular depolarization, (iv) QT interval: heart repolarization, and (v) heart rate, corresponding to the cardiac frequency (bpm). Moreover, ECG traces allowed for a qualitative analysis of heart rate changes (arrhythmias). We individually assessed the relations between the QT interval and the RR interval to obtain physiologically relevant values for heart rate-corrected QT interval (QTc) by giving a normalized RR interval (RR100=RR0/100ms). Next, the value of the exponent (y) in the formula QT0=QTc×RRy100 was assessed, in which QT0 is the observed QT and both QT and QTc are given in milliseconds (ms). By taking the natural logarithm of each side of the formula (QT0)=In(QTc)+yln(RR100), the slope of the linear relation between the log-transformed QT and RR100 thus defined the exponent to which the RR interval ratio should be raised to correct the QT for the heart rate.32
Indirect immunofluorescenceSamples of cardiac tissue, from both noninfected and T. cruzi-infected mice, were frozen in Tissue-Tek O.C.T Compound (Sakura, Zoeterwoude, The Netherlands) and stored in liquid nitrogen. Heart cryosections were fixed in cold acetone, air-dried and stored at -20°C. Sections were washed in PBS and then blocked with PBS containing 4% serum albumin bovine (BSA; PBS+BSA) before immunostaining. The samples were incubated overnight at 4°C with anti-fibronectin (1:600, Sigma Chemical Co., St Louis - USA) or type 1 Collagen (1:400; Life Technologies Co., Carlsbad, CA - USA) antibodies followed by washing with PBS+BSA. The antigen-antibody complex was detected by incubation for 1h at 37°C with TRITC-conjugated secondary antibody (1:400, Sigma). After washing, the sections were incubated with 4′,6-diamidino-2-phenylindole (DAPI), a DNA staining, and mounted with the anti-fading DABCO® (Sigma). Images were acquired under a Zeiss Axio Imager M2 fluorescence microscope using AxioVision (Zeiss) digital image processing software.
For the type I collagen bioimage analysis, at least five different fields were captured from each group (uninfected and infected), and fluorescence images were segmented and the mean intensity measured using Knime workflow.33
Protein extraction and immunoblotting assaysNoninfected and T. cruzi-infected cardiac tissue fragments were extracted with lysis buffer (Tris 50mM, NaCl 150mM, Triton X-100 1%, pH 8,0) containing protease ([4-(2-Aminoethyl)-benzenesulfonyl-fluoride hydrochloride (AEBSF), aprotinin, bestatin, Antipain-hydrochloride, E-64–[N-(trans-Epoxysuccinyl)-l-leucine 4-guanidinobutylamide, Leupeptin hemisulfate salt, Pepstatin A; Sigma Chemical Co.) and phosphatase (Cantharidin, (–)-p-Bromolevamisole oxalate and Calyculin A; Sigma) inhibitors. A small aliquot of the cell lysate (25μL) was separated for protein dosage, using the Follin–Lowry method, and the loading buffer (62.5mM Tris–HCl, pH 6.8; 2% SDS; 10% glycerol; 0.01% bromophenol blue; and 5% β-mercaptoethanol) added to the rest of the protein extract followed by heating for 5min at 100°C. A total of 20–40μg of protein was resolved by SDS–PAGE (10%) according to the antigen analyzed. The proteins were transferred to nitrocellulose membranes (BioRad, Hercules, CA) in transfer buffer (25mM Tris–HCl 192mM glycine and 10% methanol, pH 8.3). Membranes were blocked for 40min with 5% skim milk in Tris-buffered saline (TBS; 50mM Tris-Cl, pH 7.5, 150mM NaCl) containing 0.05% Tween 20 (TBST) prior to an overnight incubation (4°C) with specific antibodies (anti-FAK and anti-ERK 1/2 antibodies (1:1000) and anti-fibronectin antibody, 1:2500), except for phosphorylated proteins whose antibodies (anti-FAK-Y397, anti-FAK-S910 or anti-pERK1/2; 1:1000) were incubated in TBST containing 1% BSA. Then, the membranes were incubated for 1h with the appropriated horseradish peroxidase-conjugated second antibody (1:20,000). The antigen-antibody complex was detected by chemiluminescence kit reagents (PIERCE; Thermo Scientific, Rockford, IL) and the membranes were exposed to Hyperfilm™ ECL (GE Healthcare). All assays were normalized by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression, used as the internal control. Densitometric analysis was performed using ImageJ quantification software to measure the relative band intensity.
RNA extraction and reverse transcriptionThe hypertrophic marker atrial natriuretic peptide (ANP) gene expression was evaluated on heart fragments using a combination of TRIzol® (Invitrogen, California, USA) and RNeasy® mini kit (Qiagen, Austin, Texas, USA) for RNA extraction. Cardiac fragments (10mg) were incubated with TRIzol® (Invitrogen) and stored at liquid nitrogen until use. Afterward, the cardiac tissue was homogenized with the Ultra-Turrax tissue Dispenser (IKA, Wilmington, USA) for 30s. After 200μl chloroform addition and vigorous homogenizing, RNA was extracted from aqueous phase using a RNeasy kit, according to the manufacturer’s recommendations. The amount of total RNA was determined by measuring the absorbance at 260nm and purity at 260/280 and 260/230nm ratios, using a Spectophotometer Pico 200 (Picodrop Ltd., Saffon Walden, United Kingdom). Then, RNA (2μg) was reverse transcribed using the SuperScript III cDNA Synthesis Kit (Invitrogen) according to the manufacturer's protocol. The cDNA concentration was measured using Qubit® ssDNA Assay Kit (Life Technologies, Eugene, Oregon, USA) and adjusted to 10ng/μL final concentration.
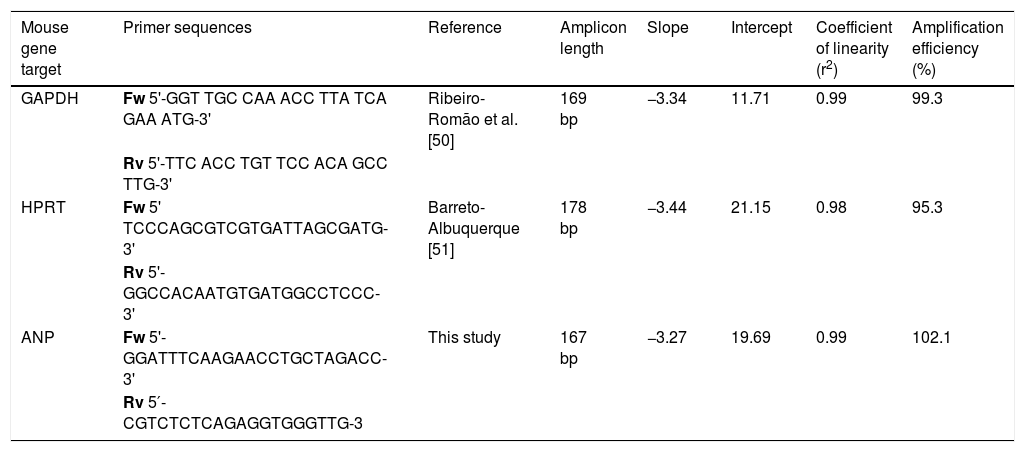
Real-time quantitative PCRANP gene expression was compared between noninfected and T. cruzi-infected cardiac tissues using Power SYBR® Green Master Mix (Life Technologies, CA, USA) RT-qPCR system. Forward and reverse primer sequences are shown in Table 1. PCR cycling conditions were: the first step at 95°C for 10min, followed by 40 cycles at 95°C for 15s and 60°C for 1min. The relative quantification of amplified products was calculated by the comparative Ct method (ΔΔCt) using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and hypoxanthine-guanine phosphoribosyltransferase (HPRT) as endogenous controls.34 The amplifications were carried out in an ABI Prism 7500 Fast device (Applied Biosystems, USA). Gene expression was expressed as fold change (2-ΔΔCt), compared to samples from uninfected mice used as calibrators. For the selection of reference gene candidates and gene expression analysis, the Expression Suite Software v1.1 (Life Technologies) was used.
Primers and standard curve parameters for hypertrophic marker amplification in C57Bl/6 mice infected with Trypanosoma cruzi.
| Mouse gene target | Primer sequences | Reference | Amplicon length | Slope | Intercept | Coefficient of linearity (r2) | Amplification efficiency (%) |
|---|---|---|---|---|---|---|---|
| GAPDH | Fw 5'-GGT TGC CAA ACC TTA TCA GAA ATG-3' | Ribeiro-Romão et al. [50] | 169 bp | −3.34 | 11.71 | 0.99 | 99.3 |
| Rv 5'-TTC ACC TGT TCC ACA GCC TTG-3' | |||||||
| HPRT | Fw 5' TCCCAGCGTCGTGATTAGCGATG-3' | Barreto-Albuquerque [51] | 178 bp | −3.44 | 21.15 | 0.98 | 95.3 |
| Rv 5'-GGCCACAATGTGATGGCCTCCC-3' | |||||||
| ANP | Fw 5'-GGATTTCAAGAACCTGCTAGACC-3' | This study | 167 bp | −3.27 | 19.69 | 0.99 | 102.1 |
| Rv 5′-CGTCTCTCAGAGGTGGGTTG-3 |
Statistical analysis and graphics were performed using R (version 3.6.0) and R Studio. Statistical difference was considered if p-value ≤ 0.05 using t-test. All RT-qPCR assays were performed with biological triplicates in two independent experiments. Results were expressed as the means±standard deviations. Statistical tests from the ΔCt values (student t-test or Mann-Whitney rank-sum test) were performed with GraphPad Prism software version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
ResultsIn an attempt to unravel the role of the FAK signaling pathway in T. cruzi cardiomyopathy, C57BL/6 mice were infected with T. cruzi Brazil strain and FAK signaling and its downstream effector (ERK1/2), acting on cardiac hypertrophy program, were analyzed on an acute and chronic infection. The peak of parasitemia (7×104 parasites/mL) was observed at 26 days post-infection (dpi) and then declined to an undetectable level after 45 dpi (data not shown). All mice survived the acute infection (30–60 dpi) and progressed to chronic infection, showing alterations in the cardiac electric conduction system observed by ECG parameters. The main electrocardiographic change was the slowing of atrioventricular (AV) nodal conduction, resulting in a prolonged PR interval (Fig. 1). A significant increase in the duration of the PR interval (p≤0.05) was observed in T. cruzi-infected mice (43.7±10.2ms) compared to uninfected groups (37.1±5.0ms), reflecting a delay in electrical conduction through AV node in chronically infected C57BL/6 mice. Another striking change was the incidence of atrioventricular blocks (AVB) in 67% of the infected mice at 150 dpi (Fig. 2). A trend towards a decrease in heart rate, or sinus bradycardia (BRA), was also observed between the groups of uninfected animals (627±42 bpm) and those infected with T. cruzi (598±140 bpm), with 50% of infected animals at 180 dpi (Fig. 2). The other parameters evaluated (P wave, QRS and QT intervals) showed no significant difference in chronically infected mice, remaining similar to uninfected animals (Fig. 1).
 Scheme of cardiac electrical impulses showing the atrial conduction (P wave), the transition between atrium and ventricle (PR interval), ventricular depolarization (QRS interval) and cardiac repolarization (QT interval). ECG analysis of uninfected (circle) and T. cruzi-infected mice (squares) demonstrated no significant difference in P wave (B), QRS (D) and QTc (E) intervals during the course of infection. However, a significant increase in the PR interval (C) and a slight decrease in heart rate (F) were evidenced in infected animals. Statistical significance relative to p≤ 0.01 between infected and uninfected mice.")
Evaluation of cardiac electrical conduction system in uninfected and Brazil strain T. cruzi-infected C57BL/6 mice. (A) Scheme of cardiac electrical impulses showing the atrial conduction (P wave), the transition between atrium and ventricle (PR interval), ventricular depolarization (QRS interval) and cardiac repolarization (QT interval). ECG analysis of uninfected (circle) and T. cruzi-infected mice (squares) demonstrated no significant difference in P wave (B), QRS (D) and QTc (E) intervals during the course of infection. However, a significant increase in the PR interval (C) and a slight decrease in heart rate (F) were evidenced in infected animals. Statistical significance relative to p≤ 0.01 between infected and uninfected mice.
 uninfected animals (NI); (B) T. cruzi-infected mice with atrioventricular block (AVB; black circle), (C) sinus bradycardia (BRA; black line), (D) ventricular extrasystole (VE; arrow) and (E) sinus arrhythmia (SA: traced line). Quantitative analysis of arrhythmia incidence in uninfected and T. cruzi-infected mice (F). A marked increase in cardiac arrhythmias was noticed during the course of T. cruzi chronic infection.")
Electrocardiographic analysis of C57BL/6 mice infected with T. cruzi Brazil strain. Heart arrhythmias typification by ECG traces: (A) uninfected animals (NI); (B) T. cruzi-infected mice with atrioventricular block (AVB; black circle), (C) sinus bradycardia (BRA; black line), (D) ventricular extrasystole (VE; arrow) and (E) sinus arrhythmia (SA: traced line). Quantitative analysis of arrhythmia incidence in uninfected and T. cruzi-infected mice (F). A marked increase in cardiac arrhythmias was noticed during the course of T. cruzi chronic infection.
When assessing the kinetics of infection, we found a low incidence of cardiac arrhythmias (20%) at the acute infection (60 dpi) with a prevalence of BRA and sinus arrhythmia (SA) among infected animal groups. Progressively, the presence of cardiac arrhythmias became more prevalent in the electrocardiographic traces, ranging from 50 to 67% from 120 dpi with BRA, AVB and supraventricular extrasystole (VE) as relevant events in chronic infection (Fig. 2).
In view of the fact that our electrocardiographic findings were indicative of heart failure, revealed by the impairment of the cardiac electrical conduction system during the chronic phase of the infection causing severe arrhythmias, such as a decrease in heart rate, sinus bradycardia and promoting cardiovascular shock, we conducted our analysis for the detection of endothelin-1 (ET-1), a hypertrophic agonist capable of activating the FAK signaling pathway. Then, we assessed the plasmatic level of ET-1 during the course of T. cruzi infection by ELISA. Elevated plasma levels of ET-1 were detected in both acute and chronic T. cruzi infections (Fig. 3). Infection times from 60 to 180 dpi showed a significant increase in ET-1 plasma level compared to the baseline level of the uninfected group (1.3 pmol/mL). Plasma levels of ET-1 increased 3.4-fold and peaked at 120 dpi, reaching a maximal value of 4.5 pmol/mL (Fig. 3).
 and chronic (90–210 dpi) phases of T. cruzi infection. A 3.4-fold increase of ET-1 level (4.5 pmol/mL) was detected at 120 dpi compared to the uninfected animals (1.3 pmol/mL). ET-1 levels were expressed as mean (± SD) pM in the plasma of infected and uninfected C57BL/6 mice. Student’s t-test: *p<0.05, **p<0.01.")
ET-1 plasma level in Brazil strain T. cruzi-infected C57BL/6 mice. Elevated ET-1 level was observed in the acute (60 dpi) and chronic (90–210 dpi) phases of T. cruzi infection. A 3.4-fold increase of ET-1 level (4.5 pmol/mL) was detected at 120 dpi compared to the uninfected animals (1.3 pmol/mL). ET-1 levels were expressed as mean (± SD) pM in the plasma of infected and uninfected C57BL/6 mice. Student’s t-test: *p<0.05, **p<0.01.
We further assessed if the abnormalities in cardiac hemodynamics and the increase of plasma ET-1 level induced activation of the FAK signaling pathway, since this protein tyrosine kinase (PTK) has been highlighted in response to changes in mechanotransduction mediated by cell stretching and cardiac overload. Our findings demonstrated the activation of FAK signaling in cardiac tissue of T. cruzi-infected mice, evaluated by phosphorylation of both tyrosine 397 (Y397) and serine 910 (S910) residues (Fig. 4). Although total FAK level was unaltered in T. cruzi-infected heart, FAK autophosphorylation at tyrosine residue 397 (Y397) was significantly increased after 90 dpi (0.9±0.2), showing an activation peak at 120 dpi (1.04±0.3) with 3.4-fold increased phosphorylated levels of FAK-Y397 (Fig. 4). A similar FAK activation curve was observed for phosphorylated FAK-S910 expression. However, high levels of FAK-S910 were noticed at 120 (1.0±0.1) and 180 (1.12±0.2) dpi, increasing 3.3 to 3.6-fold, respectively, compared to standard activation levels of uninfected animals (0.3±0.08) (Fig. 4). Interestingly, FAK activation correlates with the ET-1 plasma level elevation and the enhancement of bradycardia (50%) in infected mice. Thus, since ERK1/2, a member of mitogen-activated protein kinases (MAP kinase) signaling pathway, has been implicated in the hypertrophic response modulated by FAK, the expression of total and phosphorylated ERK1/2 (pERK1/2) were also analyzed. Total ERK1 expression was practically invariable, except for the decline at 210 dpi, while its level of phosphorylation increased throughout the course of the infection (60–210 dpi) (Fig. 4). An interesting fact is that the activation of ERK1/2 occurred prior to the activation of FAK, but coincided with the ET-1 plasma level increases. Phosphorylated ERK1/2 (pERK1/2) level peaked at 180 dpi (4.7±0.7), coinciding with the FAK-S910 peak (Fig. 4).
 Representative immunoblot images of FAK, FAK-Y397 and FAK-S910. (B) An increase in FAK autophosphorylation (FAK-Y397) in the cardiac tissue occurred at the early chronic infection (90 dpi) and peaked at 120 dpi. (C) High levels of phosphorylated FAK-S910 was noticed from 120 to 180 dpi without alteration in total FAK expression (D). (E) Representative immunoblot images of ERK1 and phospho-ERK1/2 (pERK1/2). (F) pERK1/2 expression increased during the course of T. cruzi infection (60–210 dpi), peaking at 180 dpi. However, no change in ERK1 expression was observed in the cardiac tissue of infected animals, except the decline in ERK1 level at 210 dpi (G). GAPDH was used as an internal loading control. Student’s t-test: ns - not significant, *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.")
Activation of FAK signaling in T. cruzi chronic infection. (A) Representative immunoblot images of FAK, FAK-Y397 and FAK-S910. (B) An increase in FAK autophosphorylation (FAK-Y397) in the cardiac tissue occurred at the early chronic infection (90 dpi) and peaked at 120 dpi. (C) High levels of phosphorylated FAK-S910 was noticed from 120 to 180 dpi without alteration in total FAK expression (D). (E) Representative immunoblot images of ERK1 and phospho-ERK1/2 (pERK1/2). (F) pERK1/2 expression increased during the course of T. cruzi infection (60–210 dpi), peaking at 180 dpi. However, no change in ERK1 expression was observed in the cardiac tissue of infected animals, except the decline in ERK1 level at 210 dpi (G). GAPDH was used as an internal loading control. Student’s t-test: ns - not significant, *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.
To address whether the cardiac hypertrophy program was triggered in response to the activation of the FAK (Y397 and S910 residues)—ERK1/2 signaling cascade, we investigated the atrial natriuretic peptides (ANP) mRNA expression, a hypertrophic marker, by RT-qPCR. Marked upregulation of ANP mRNA expression was revealed in the myocardium of infected mice. Our results demonstrated that the ANP mRNA level tended to increase at 120 dpi, but a significant 2-fold increase in the expression of this hypertrophic marker was noticed in the Brazil strain infected mice from 150 dpi onwards (Fig. 5).
. HPRT and GAPDH genes were used as endogenous controls. The mean Ct values between control and infected groups were evaluated using Student’s t-test or non-parametric Mann-Whitney rank sum test. *p≤0.05, **p≤0.01.")
Trypanosoma cruzi chronic infection induces hypertrophic gene expression. Increased ANP mRNA expression, evaluated by RT-qPCR, was observed in the myocardium of C57BL/6 mice chronically infected with T. cruzi (150–210 dpi). HPRT and GAPDH genes were used as endogenous controls. The mean Ct values between control and infected groups were evaluated using Student’s t-test or non-parametric Mann-Whitney rank sum test. *p≤0.05, **p≤0.01.
Additionally, the remodeling of the cardiac extracellular matrix (ECM) has been shown to be associated with myocardial hypertrophy and heart failure.35 Thus, the myocardial fibrosis profile was also analyzed in the myocardium of T. cruzi-infected C57BL/6 mice. The spatial distribution and ECM components expression, as type 1 collagen (COL1) and fibronectin (FN), were assessed in the cardiac tissue. A thin deposition of COL1 (Fig. 6) and FN (Fig. 7) was observed in the myocardial interstitium of uninfected animals. A slight increase in COL1 deposition, as revealed by fluorescence images, was observed in the infected tissue at 60 and 90 dpi, becoming a dense fibrillar COL1 fluorescence signal at a later time of infection (120–210 dpi) (Fig. 6). The quantitative analysis, using Knime bioimage tools, also confirmed the augment in the COL1 expression in the myocardium of the infected mice (Fig. 6). Likewise, FN spatial distribution analysis also revealed an intense fluorescent signal, mainly after 60 dpi, with a dense FN deposition in the infected cardiac tissue (Fig. 7). Infected animals also showed enhancement in FN level, demonstrated by immunoblotting assay, showing a significant increase in FN expression from 60 dpi compared to cardiac tissue from uninfected mice (Fig. 7).
 expression in the myocardium of T. cruzi-infected mice. (A) Distribution of COL1 (red) in the cardiac tissue of uninfected mice. A gradual increase in COL1 deposition was noticed in the myocardium at later acute (60 dpi; B) and early chronic infection (90 dpi; C). Dense COL1 fibrils were clearly seen at 120 (D), 150 (E), 180 (F) and 210 dpi (G). The increase in COL1 expression was confirmed by measuring the COL1 fluorescence signal with Knime workflow (H). DAPI (blue) stained nucleus. Bar=10μm. Student’s t-test: **p≤0.01; ***p≤0.001; ****p≤0.0001.")
Enhancement of type 1 collagen (COL1) expression in the myocardium of T. cruzi-infected mice. (A) Distribution of COL1 (red) in the cardiac tissue of uninfected mice. A gradual increase in COL1 deposition was noticed in the myocardium at later acute (60 dpi; B) and early chronic infection (90 dpi; C). Dense COL1 fibrils were clearly seen at 120 (D), 150 (E), 180 (F) and 210 dpi (G). The increase in COL1 expression was confirmed by measuring the COL1 fluorescence signal with Knime workflow (H). DAPI (blue) stained nucleus. Bar=10μm. Student’s t-test: **p≤0.01; ***p≤0.001; ****p≤0.0001.
 expression in chronically infected C57BL/6 mice. (A) FN spatial distribution in the myocardium of uninfected mice. (B–G) Thick deposition of FN was observed in the cardiac tissue of T. cruzi-infected mice. Intense fluorescence signal in the myocardium interstitium was observed at 60 (B), 90 (C), 120 (D), 150 (E), 180 (F) and 210 dpi (G). Representative immunoblotting image and densitometric analysis showing a significant increase of FN expression in acute (60 dpi) and chronic infection (90–210 dpi) (H). DAPI (blue) stained the nucleus. Bar=10μm. GAPDH was used as an internal control for protein normalization. Student’s t-test: *p≤0.05; **p≤0.01; ***p≤0.001.")
Fibronectin (FN) expression in chronically infected C57BL/6 mice. (A) FN spatial distribution in the myocardium of uninfected mice. (B–G) Thick deposition of FN was observed in the cardiac tissue of T. cruzi-infected mice. Intense fluorescence signal in the myocardium interstitium was observed at 60 (B), 90 (C), 120 (D), 150 (E), 180 (F) and 210 dpi (G). Representative immunoblotting image and densitometric analysis showing a significant increase of FN expression in acute (60 dpi) and chronic infection (90–210 dpi) (H). DAPI (blue) stained the nucleus. Bar=10μm. GAPDH was used as an internal control for protein normalization. Student’s t-test: *p≤0.05; **p≤0.01; ***p≤0.001.
A plethora of signaling pathways have been implicated in the pathophysiology of cardiac hypertrophy. Thus, delineating the cellular and molecular mechanisms underlining the chagasic cardiac hypertrophy sheds light on Chagas disease pathogenesis. In this study, we addressed the role of FAK signaling in T. cruzi induced cardiac hypertrophy and heart failure.
FAK signaling emerged as a key cardiac hypertrophic regulator in response to biomechanical stress and humoral stimuli.36 Our data suggest that both mechanical and hypertrophic agonist stimuli are involved in T. cruzi cardiac hypertrophic response. Integrin, a transmembrane receptor involved in force transmission, connects the contractile muscle apparatus to extracellular matrix components (ECM), regulating the mechanical force. FAK, a key molecule in mechanosignalling, interacts with integrin through structural proteins, as talin and vinculin, in focal adhesion sites and costameres.37 Pressure overload induces FAK tyrosine phosphorylation and promotes c-Src activation in addition to downstream effectors of FAK, as ERK1/2 and AKT via GRB2 and phosphatidylinositol 3-kinase (PI3K).21,26,38,39 Our findings demonstrated that changes in integrin-mediated mechanical force, induced by fibrosis leading to pressure overload, activates FAK signaling and its downstream effector involved in hypertrophic response. The abnormal ECM deposition, revealed by enhancement of COL-1 and FN expression in cardiac tissue, causes its stiffness and disturbs mechanical homeostasis, altering integrin-mediated mechanotransduction. Additionally, disruption of vinculin costameres in T. cruzi infected myocardium may also contribute to alterations in the transmission of cardiac contraction force.40 In this scenario, elevated FAK-Y397 phosphorylation level coincides with the ECG abnormalities, showing reduced heart rate, prolonged PR interval and increased rate of arrhythmias (from 90 dpi) suggestive of cardiac hypertrophy and heart failure. Interestingly, an in vitro model of acute infection by T. cruzi with high parasitic load revealed loss of spatial organization and downregulation of mechanosensitive proteins in cardiomyocytes, such as talin and paxilin, inducing a downregulation of the FAK signaling pathway activation and therefore, suggesting a disruption of integrin-dependent signaling transduction.41 FAK signaling activation has also been shown to be a beneficial event in ischemia/reperfusion, promoting cardiomyocytes survival under stress conditions. Activation of the NF-kB survival signaling pathway, as downstream of FAK activation, modulates FAK-dependent cardioprotection by reducing pro-apoptotic genes level.42
In addition to integrin-regulated FAK activation, this tyrosine kinase is also activated by ET-1, the hypertrophic agonist. ET-1, a potent vasoconstrictor involved in Chagas disease pathogenesis, has been reported to trigger FAK activation and promotes cardiac hypertrophic remodeling.43 As expected, Brazil strain C57BL/6 mice infection induced ET-1 plasma level increase and cardiac dysfunction in both acute and chronic infection. High ET-1 plasma level has been reported in patients with Chagas cardiomyopathy44 and also in a mouse model of T. cruzi chronic infection (Brazil strain), which evolved to right ventricular enlargement.15,19 In our findings, elevated ET-1 levels were associated with ERK1/2 phosphorylation up-regulation, still in the acute phase of infection (60 dpi). Also, increased ERK1/2 activation preceded FAK-S910 phosphorylation (120 dpi), suggesting that FAK undergoes FAK-S910 phosphorylation via ERK1/2 signaling as previously reported for dilated cardiomyopathy (DCM).20 ET-1stimulated neonatal rat ventricular myocyte (NRVM) cultures, in an ETAR-dependent protein kinase C delta (PKCδ) activation, induced ERK1/2 activation through Raf-1—MEK1/2 effectors, resulting in FAK-S910 phosphorylation and sarcomere assembly during cardiac myocyte hypertrophy.20 Also, bi-directional crosstalk between Src and PKCδ has been proposed with ET-1stimulating PKCδ-MEK1-ERK1/2 and Src-MEK1-ERK1/2 pathways20,45 or Src-MEK5-ERK5.20 Sustained MEK1-ERK1/2 activation has been shown to promote cardiac hypertrophy and anti-apoptotic effect.46 Additionally, ET-1 has also been highlighted in T. cruzi infection due to its role in triggering the Ca2+/calcineurin (Cn)/nuclear factor of activated T cells (NAFT) signaling pathway, causing the activation of fetal gene program.16 C57BL/6 and Balbc mice infected with T. cruzi Y strain showed high levels of ET-1 and eicosanoids (cyclooxygenase-2; COX2) associated with high tissue parasitism and myocarditis. The cooperative action between ET-1 and T. cruzi infection regulates the increase in COX2 expression by activating the Cn/NAFTc4 signaling pathway in HL-1 atrial myocytes, leading to ANP production.16 Curiously, our results demonstrated a significant increase in ANP mRNA expression only from 150 dpi, but with a tendency to increase by 120 dpi. Although hypertrophy has been well established in Brazil strain infected C57BL/6 mice, this experimental model of T. cruzi infection gradually develops dilated cardiomyopathy with an increase in right ventricular internal diameter after 100 dpi.15 In contrast, a study analyzing the electrocardiographic profile of different mice lineage and T. cruzi strains, as a model for experimental chagasic cardiomyopathy, did not show significant changes in the ECG of B6 mice infected with Brazil strain, probably due to the short period of infection analyzed (up to 13 weeks).47
Activation of hypertrophic response program in response to T. cruzi infection has been shown to depend on Toll-like receptor 2 and NF-kB activation, leading to an increase in the IL-1β level and cardiomyocyte hypertrophy (Petersen et al., 2005).11 On the other hand, PI3K-AKT/nitric oxide (NO) signaling has been demonstrated to be implicated in the cardiac myocyte remodeling of chagasic cardiomyopathy.12 C57BL/6 mice infected with Colombian strain induced PI3K-AKT/NO signaling pathway activation with cardiac hypertrophy evidenced in the acute (30 dpi) and the early chronic phase (90 dpi) of T. cruzi infection, showing increased mRNA level of hypertrophic markers.12 A remarkable feature is that ERK1/2 and AKT have been highlighted as a downstream effector of FAK activation37,48 and FAK signaling may also regulate NF-kB activation,49 suggesting that chagasic cardiac hypertrophy may be regulated by complex regulatory interactions involving interlaced networks of multiple signaling pathways.
ConclusionOur data contribute with new insights on the regulation of cardiac hypertrophy induced by T. cruzi infection. We demonstrated that the FAK signaling pathway regulates the hypertrophic process in chronic chagasic cardiomyopathy. Cardiac overload induced by ET-1-mediated extracellular matrix remodeling and tissue damage triggers the hypertrophic response via ET-1-ERK1/2-FAKS910 and integrin-FAKY397-ERK1/2, leading to activation of the fetal gene program, heart failure and hypertrophy. Further studies will be carried out to evaluate the effect of FAK signaling inhibitors in the therapy of Chagas disease.
Conflicts of interestThe authors declare no conflicts of interest.
This work was supported by grants from the Oswaldo Cruz Institute of the Oswaldo Cruz Foundation (Fiocruz), Programa Estratégico de Apoio à Pesquisa em Saúde (Papes VI)/Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil (grant 421856/2017-3 and 424015/2018-8), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (grant E-26/010.101050/2018) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001. The authors thank Alanderson da Rocha Nogueira for technical support and the Platform of Real Time PCR RPT09A from Fiocruz, by the use of the equipment and support.
