





Leishmania major is a causative agent of zoonotic cutaneous leishmaniasis in the center of Iran, Abarkouh district. Molecular characterization and precise incrimination of Leishmania species was carried out to perform controlling measurements and to design treatment programs for zoonotic cutaneous leishmaniasis.
MethodsAll smears isolated from ulcers of suspected patients were examined under a light microscope and graded for amastigotes frequency. Extraction of DNA, PCR, RFLP and sequencing of ITS-rDNA genotype were done to increase the efficacy of Leishmania parasites identification at their species-specific level and to detect any Leishmania infections within.
ResultsHumans were found to be infected with L. major with high infection frequency and also Leishmania tropica was identified with low occurrence for the first time as non-native species using molecular analyses. The rates of infections was considerable with microscopic observation (n = 65, 73%) out of 89 smears prepared from suspected patients. Molecular analyses showed that the density of L. major was significantly higher (n = 48, 53.93%) than L. tropica (n = 4, 4.49%) (Mann–Whitney U test: p < 0.05) and two samples (2.25%) remained ambiguous after several sequencing. L. major did not have diversity with two common haplotypes but L. tropica were found to exhibit high diversity with three novel haplotypes.
ConclusionL. major was considered the causative agent of leishmaniasis in the region, but the identification of a non-native L. tropica revealed the importance of further isolation of Leishmania parasites following molecular analyses and confirmation, and also revealed the importance of further isolation of Leishmania parasites from patients of the field areas who do not have easily access to health care centers for specialized treatment strategies.
Cutaneous leishmaniasis (CL) is a noticeable tropical disease which has lots of adverse effects on human health in 98 countries on 5 continents in the world as well as Iran.1,2 The mammalian host(s) of CL in the Old World including Rodentia, Carnivora and human has been infected by different species of single-celled Leishmania parasites. Humans are naturally infected by the bite of female sand flies from vertebrate animals in the form of zoonotic cutaneous leishmaniasis (ZCL) with the exception of Leishmania tropica which is often known as an anthroponotic transmissible disease from humans to vertebrate animals.3,4 An epidemiological study of ZCL in 11 provinces of Iran showed that L. major was the causative agent of CL in rural areas of Iran with 95% prevalence rate whereas L. tropica had a distribution rate of 65% in urban districts.4 Although, papers have been published on the transmission and epidemiological circulation of Leishmania parasites in vectors,5 reservoir hosts6,7 and humans from different ZCL foci of Iran,2,8 there is no considerable and sufficiently investigation on leishmaniasis in humans at the center of Iran (Yazd province). Nevertheless ZCL is considered as an endemic disease in Isfahan province adjacent to Yazd, it has also become endemic in several areas in Yazd province at the center of Iran during the last 10 years.9,10 Therefore, the constant appearance of infectious agents of Leishmaniasis within a human population of this focus highlights the direct significant attention to identifying transmission cycles of Leishmania parasites and their clinical manifestations in humans of Yazd district.
Leishmania major is one of the leading-off agents causing rural, zoonotic and vector-borne disease and the digenetic form of Leishmania life cycle is completed in different species of wild rodents and phlebotomine sandflies as reservoir hosts and vectors respectively in many geographical locations where it occurs.10 In fact L. major is known as the causative agent of ZCL and more frequent than the other Leishmania parasites in Iran which induces Th2 immune response in case of exacerbation with disfigured cutaneous patterns.11 In addition, another principal agent of cutaneous leishmaniasis, L. tropica, has been implicated in occasional cases of recidivans or viscerotropic cases.12 Recently, L. tropica has stimulated many interests because of its potential traits to visceralize and/or classical visceralize in humans,13 disseminating of cutaneous leishmaniasis along with visceral leishmaniasis14 and developing of mucosal leishmaniasis in Iran.15 An epidemiological study of ZCL in 11 provinces of Iran.15 Accordingly, finding non-native species of L. tropica is of great importance in studied region.
However, three species of Leishmania parasites have been incriminated as the causative agents of human leishmaniasis in Iran. They are L. major, L. tropica and L. infantum.16 Also, some other mammals’ Leishmania such as L. turanica, L. gerbilli and L. close to gerbilli have been reported from Iran.2 and isolated from reservoir hosts and sand flies vectors in different CL foci of Iran.6,16
The popular and practical method for identification of Leishmania species is Giemsa stained smears prepared from patients’ ulcer with the examination of Leishmania amastigotes under microscopic observation.17 In addition, incrimination of Leishmania parasites was previously based on clinical symptoms, vector assessments, and pathogenicity in laboratory animals and growth in media.9,18 In recent years, Leishmania species have firmly been identified because of high sensitivity, more rapid determination and characterization after amplifying targeted genes using PCR, RFLP and sequencing.15,19
In our investigations, PCR amplification, RFLP digestion and sequencing of ITS-rDNA genotype were employed to identify all Leishmania species isolated from the lesion of suspected patients having serous exudate.
The motivation of this work arises from increasing problems of leishmaniasis in the center district of Iran. In fact, this investigation was conducted to monitor the effectiveness of control measures, to accurate incrimination of Leishmania parasites at their species level with molecular analyses, to characterize the species-specific parasites isolated from humans residing in the natural field working regions and also to improve our knowledge about all Leishmania species those readily maintain their ecological circulation in the endemic focus of Yazd province.
MethodsOrigin and sampling of Leishmania parasites from suspected patientsWithin the ZCL focus, prepared samples of suspected patients were collected from 16 surveyed villages of Abarkouh city in Yazd province (Fig. 1). Yazd district is geographically located between 31° 07′ 44″ N and 53° 16′ 57″ E of central Iran. The city proper is situated at an altitude of 1510 m (4,954 ft) above sea level (a.s.l.) and all villages of this region were screened and sampled from suspected patients from 2014 to 2016. The smears firstly isolated from patients’ lesion, prepared on the slide (slide fixation) and froze within the days of collection. Samples were then transferred on ice to the Pasteur Institute of Iran, Tehran, for microscopic identification and molecular experiments.
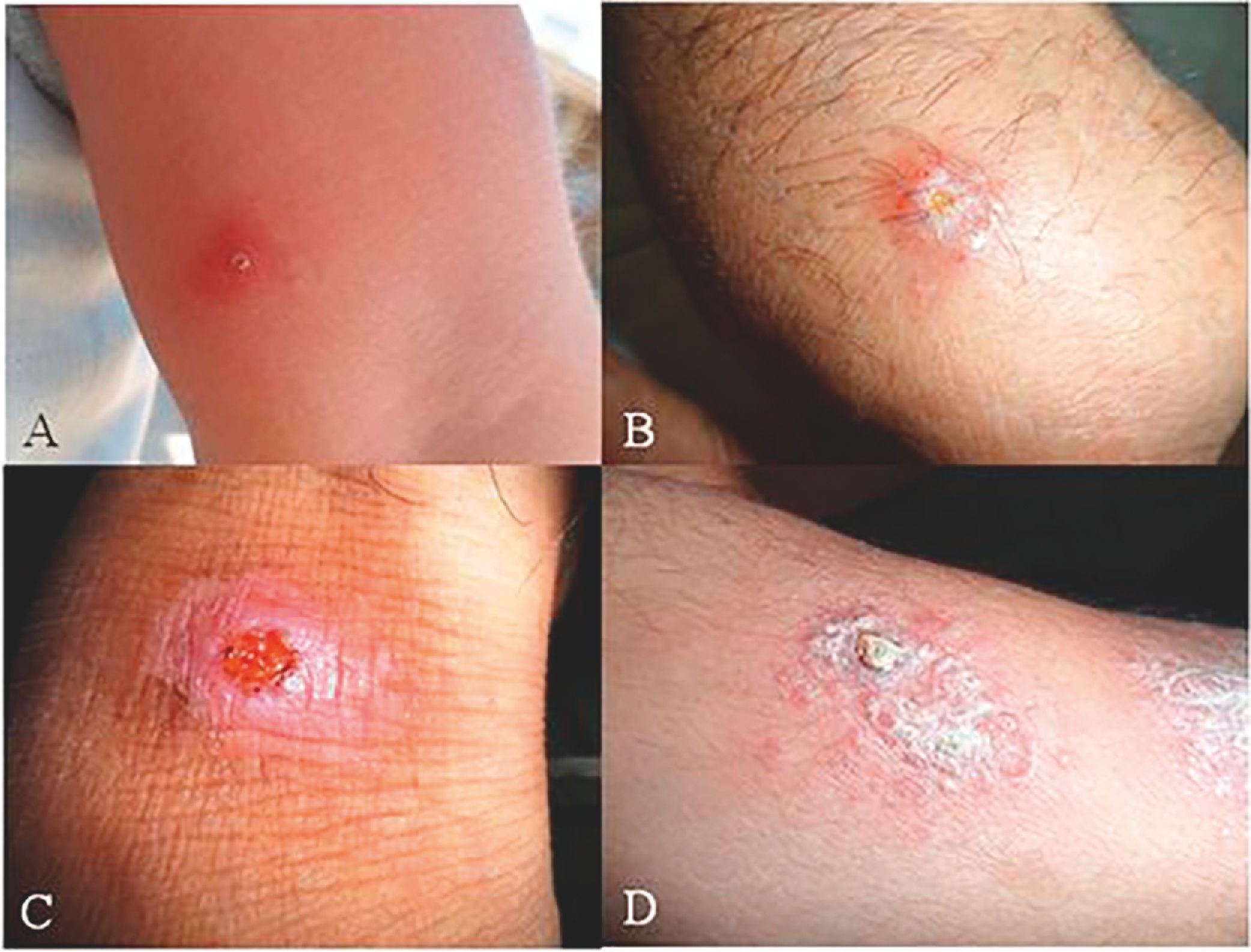
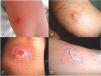
The occurrence of leishmaniasis was very low in four consecutive months (May–August). Whereas we found a sharp increasing rate of CL lesions during the 3rd Quarter 2014 (October–December), sampling was carried out at the time of lesions’ appearance in Abarkouh district (Fig. 1). The smears were prepared from the active lesions of patients residing in different rural regions of leishmaniasis throughout Abarkouh district. The personal information, lesion duration, type and the number of lesion, ulcer(s)’ location, patients’ travelling to endemic areas and also drug consumption were recorded for each patient separately. Expanded set of tools (conventional and then molecular methods) were exerted to recognize samples including presumptive CL parasites based on Evans protocol17 and were then smeared on two microscopic slides, air dried, fixed with methanol and stained by Giemsa. All collected smears were examined under a light microscope (Nikon YS100) with high magnification (1000×) and identified to Leishmania infection microscopically (Fig. 2). The sizes of amastigotes and different morphometric shapes of leishman bodies of Leishmania parasites were precisely evaluated (30 min. per slide) by ocular micrometer (×100 objective = 1 µm per unit space) and Dino Capture 2.0. The positive smears of each patient were scored for Leishmania amastigotes density and frequency from 1+ to 6+.2 The distribution of our statistical population was asymmetric and not normal; therefore we required using a non-parametric statistical Mann–Whitney U test in SPSS 16: p < 0.05.
DNA extraction of genomic DNA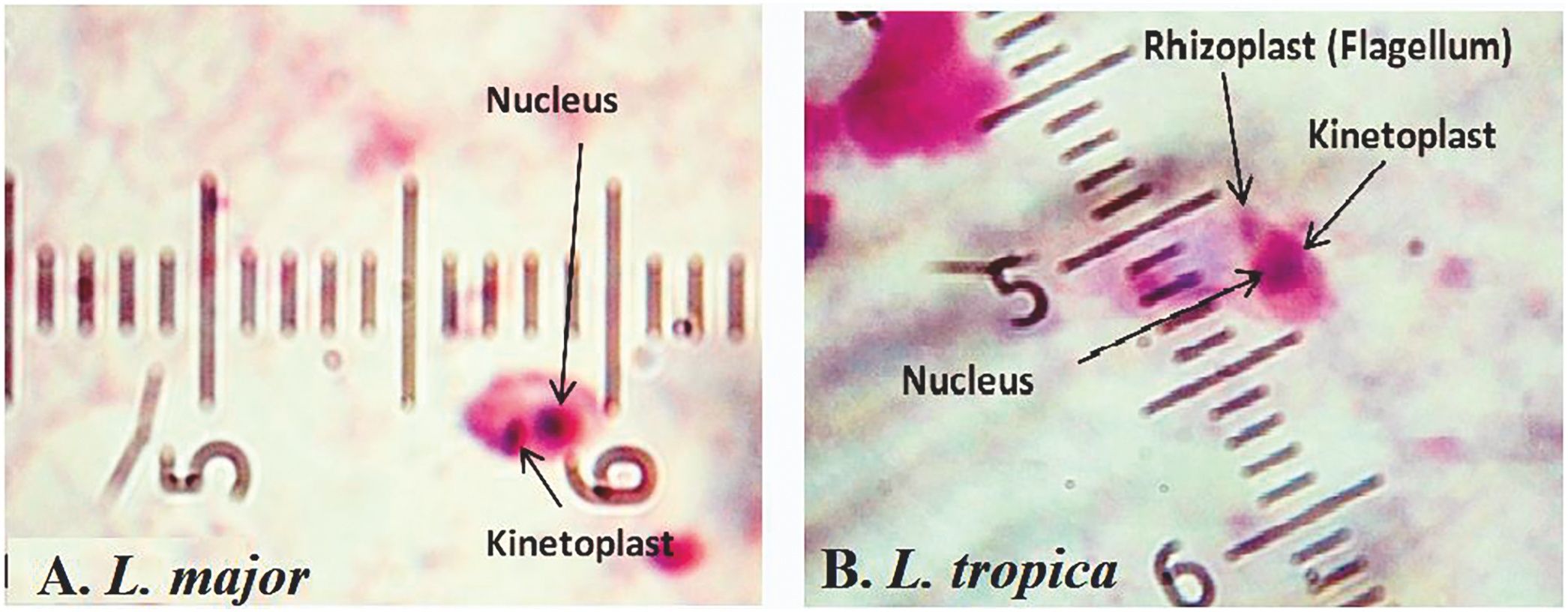
All Giemsa-stained slide washed with absolute ethanol for eliminating immersion oil on the smears and covered with 300 μl lysis buffer (50 mM NaCl, 50 mM Tris, 10 mM EDTA, pH 7.4, 1% (v/v) Triton x-100) for the extraction of total DNA from graded slides. After a short time the smears completely removed and transferred to a 1.5 ml reaction tube. Then, 15 µl proteinease K (20 mg/ml, fermentase) was added to each tube. Cell lyses was accomplished after incubation for 4–6 h at 56 °C. The lysate was extracted by phenol–chloroform followed by ethanol precipitation.18
The DNA was re-suspended in 30 μl TE1X and stored at −20 °C. The genomic DNA of each smear from suspected patient and any parasite within was extracted in the Molecular Systematics Laboratory suite in a room where amplified and cloned DNAs were never processed.
PCR amplification and molecular identification of Leishmania parasitesEach PCR assay was executed to screen Leishmania infection by the nested PCR of a fragment of ITS (nuclear ribosomal) gene. For the first stage of nested PCR, forward primer IR1 with the reverse primer IR2 and for the second-stage forward primer ITS1F with the reverse primer ITS2R4 were used.16 Double distilled water and DNA from L. major and L. tropica were used as negative and positive controls for each batch of PCR (primers were produced at CinnaGen Co.). The reactions were performed in an Eppendorf Mastercycler Gradiant device. PCR products were subjected to electrophoresis in 1.5% agarose gel at 150 V in 0.5 × TBE (0.045 M Tris-borate,1 mM EDTA) (Merck Co.) buffer and monitored under ultraviolet light after staining for 15 min with (0.5 g/ml) safe stain; in order to see an approximately 480 bp (including primers) showing ITS-rDNA gene fragment for Leishmania infections. The reactions were performed in an Eppendorf Mastercycler Gradiant device.
Identification of the species described by the molecular analyses (RFLP and sequencing)To select the suitable enzyme for digesting the PCR product in RFLP method, the sequences of standard strains (L. major, L. tropica, L. infantum, L. turanica and L. gerbilli) registered in GenBank were employed for sequence analysis. Standard strains sequencing of Leishmania were examined by in silico analysis, CLC DNA Workbench 5.2 software (CLC bio A/S, Aarhus, Denmark). A suitable restriction enzyme BsuRI (HaeIII) (ParsGene Co.) was selected with different cut sites GG↓CC as an appropriate restriction enzyme digestion to incriminate different CL species.
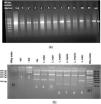
Endonuclease digestion was done in a volume of 30 μl consisting of 10 μl of PCR product, 2 μl of BsuRI (HaeIII) enzyme (Fermentas), 2 μl of 10× buffer and 16 μl of distilled water for 4 h (rapid digestion) at 37 °C for ITS-rDNA gene fragment (Fig. 3). Also, standard strains of WHO were employed as positive controls as fallow: L. major (MHOM⁄SU⁄73⁄5ASKH), L. tropica (MHOM/PS/2008/344Jn SF53) and L. donovani (MHOM⁄TN⁄80⁄IPT1). Two negative controls were utilized, one of which did not have restriction enzyme and the other one did not have PCR product. After endonuclease digestion of PCR product with BsuRI (HaeIII) enzyme, the fragments were analysed using electrophoresis on agarose gel 3% containing safe stain and ladder 50 bp (Fermentas) (Fig. 3).
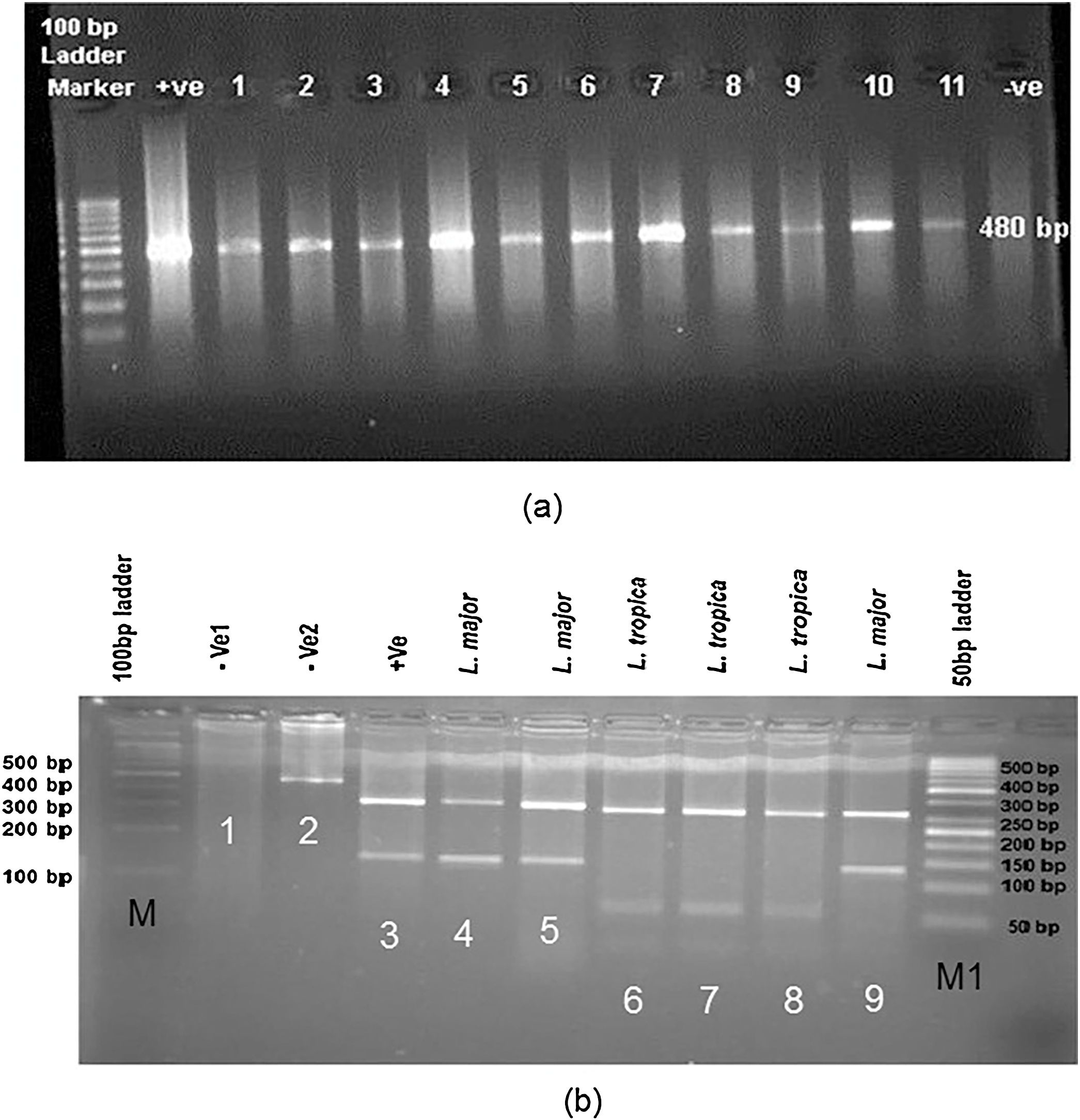
(a) Leishmania positive results isolated from freshly ulcers of suspected patients by PCR targeting ITS-rDNA genotype without RFLP. (b) PCR-RFLP observation in Yazd isolates based on ITS-rDNA genotype. Lane 1 (−ve, negative control containing BsuR1 without PCR product), lane 2 (−ve: negative control containing PCR product without BsuR1), lane 3 (+ve, positive control for L. major), lanes 4, 5; Yaz32 and Yaz19 (L. major, common haplotypes): isolated from wet lesions of suspected patients in Esfand-Abad and Chahgir, respectively. Lanes 6–8; MH488992 (Yaz58), MH492002 (Yaz60), and MH488993 (Yaz61) (L. tropica, three Novel haplotypes): isolated from dry lesions of suspected patients western and southern Yazd, Lane 9; Yaz27; L. major isolated from wet lesion of suspected patient, central Yazd. M, 100 bp size marker; M1, 50 bp size marker; –ve, negative; +ve, positive.
To reconfirm the results of PCR-RFLP, the numbers of PCR products were sequenced by targeting ITS-rDNA in both directions using the ABIPRISMTM 3130 Genetic Analyzer automated sequencer (Applied Biosystem, USA). Individual sequences were justified, edited and aligned in consensus positions with GenBank sequences of all regional species in case of homology and similarity. The Sequencher™v. 4.1.4 Software for PC was used to incriminate unique haplotypes (PC, Gene Codes Corporation). The MEGA v5.05 software was applied for phylogenetic analysis.20 To permit grouping of the new species, the phylogenetic maximum likelihood (PhyML)21 program was run to get maximum likelihood tree based on the Kimura 2-parameter model of nucleotide substitution search by stepwise addition of 100 random replicates, and nonparametric ML bootstrap was calculated with 1000 replicates.
ResultsMicroscopic observation and molecular experimentsThe presence of L. tropica was proved as a non-native species where there was no report of this species before. To acquire an appropriate statistical results, all patients (n = 89) having distinctive, fresh, acute and/or chronic lesions (wet, dry and/or mixed) examined in all 16 important villages from different sites of Abarkouh (12500 populations) (Fig. 1). L. major and L. tropica were firmly found and molecularly typed from the active lesion of patients. L. major was identified with regular amastigotes’ shapes (round or oval with a size of 2–4 µm) whereas appearances of L. tropica amastigotes were irregular (pear, spindle or cigarette with more than 4 µm) (Fig. 2). Infections were found from the lesion of individuals (ranging from <1 to >25), residents of villages (Abarkouh region) who had L. major more in face, hand and foot isolated from wet lesion respectively and, L. tropica in face and hand isolated from dry lesion respectively (Table 1). Of 89 samples, 65 (73%) Giemsa stained slides were observed positive microscopically for Leishmania amastigotes and the specified score was designated to each prepared slide smear (Table 1). Results showed that the median prevalence of ZCL was considerable in Abarkouh district (approximately 71.2 cases per 10,000 inhabitants). All 65 Leishmania positive samples were applied to screen by PCR assay that only 54 samples were successfully identified Leishmania positive using ITS-rDNA gene fragment and then subjected to RFLP following sequencing. To identify species-specific Leishmania parasites, molecular experiments were carried out on all microscopically observed grading numbers (n = 65) using PCR. The positive PCR samples are as follows: 1+ (n = 6 out of 15), 2+ (n = 5 out of 7), 3+ (n = 14), 4+ (n = 14), 5+ (n = 11) and 6+ (n = 4). This inclusive inquiry revealed that the number of patients those who had Leishmania lesions were more in Chahgir (n = 17) and Abarkouh (n = 16) than the other villages. The samples of +1 grading number exhibited (n = 15, 16.8%) at the highest rate and the +6 grading number remained at the lowest rate (n = 4.5%) isolated from the suspected patients of collected sites (Table 1).
Microscopic observations; MO, on Giemsa-stained slides prepared from human lesion and molecular analyses on Leishmania positive of MO using ITS-rDNA genotype, Abarkouh district, Yazd province.
| Collection site | Total samples | Microscopic observation | Personal information based on age (years) and lesion type | Molecular analysis | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Province | District | Villages of Abarkouh | Positive grading numbers of Leishmania amastigotes based on Giemsa-stained smears | Sex | <1 | 1-5 | 5-10 | 10-15 | 15-25 | >25 | PCR using ITS-rDNA genotype + ve | RFLP with BsuRI & Sequencing | |||||||||||||||||
| +ve (%) | −ve (%) | Total +ve | F | M | W | D | W | D | W | D | W | D | W | D | W | D | L. major +ve | L. tropica +ve | NI +ve | ||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | ||||||||||||||||||||||||
| Yazd | Abarkouh | Chahgir | 17 | 2 | 0 | 1 | 4 | 2 | 0 | 8 | 9 | 9 | 8 | 1 | 1 | 2 | 3 | 5 | 1 | 4 | 8 | 6 | 2 | 0 | |||||
| Nosrat Abad | 6 | 2 | 1 | 1 | 1 | 1 | 0 | 0 | 6 | 1 | 5 | 1 | 1 | 3 | 1 | 4 | 3 | 0 | 1 | ||||||||||
| Taghi Abad | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 2 | 2 | 1 | 1 | 0 | 0 | |||||||||||||
| Esfand Abad | 14 | 4 | 0 | 2 | 3 | 2 | 0 | 3 | 11 | 6 | 8 | 1 | 1 | 2 | 4 | 4 | 2 | 10 | 9 | 1 | 0 | ||||||||
| Ardi | 3 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 3 | 0 | 3 | 1 | 2 | 2 | 1 | 0 | 1 | ||||||||||||
| Asad Abad Sofla | 4 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 3 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 0 | |||||||||||
| Mehr Abad | 8 | 0 | 3 | 1 | 1 | 0 | 1 | 2 | 6 | 2 | 6 | 1 | 1 | 1 | 3 | 2 | 6 | 6 | 0 | 0 | |||||||||
| Harooni | 7 | 0 | 0 | 1 | 0 | 2 | 2 | 2 | 5 | 3 | 4 | 1 | 1 | 1 | 1 | 1 | 2 | 5 | 5 | 0 | 0 | ||||||||
| Asad Abad-Esfand Abad | 2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 2 | 2 | 1 | 1 | 0 | 0 | |||||||||||||
| Hossein Abad | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | |||||||||||||
| Raeis Abad | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | ||||||||||||
| Kolpeh | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | |||||||||||||
| ShahreAsb | 4 | 1 | 0 | 2 | 1 | 0 | 0 | 0 | 4 | 1 | 3 | 2 | 2 | 3 | 3 | 0 | 0 | ||||||||||||
| Harook | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | |||||||||||||
| KhoramAbad | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | |||||||||||||
| Abarkouh | 16 | 2 | 2 | 4 | 1 | 1 | 0 | 6 | 10 | 4 | 12 | 1 | 2 | 3 | 3 | 1 | 3 | 1 | 2 | 9 | 9 | 0 | 0 | ||||||
| Total (%) | 89 | 15(16.8) | 7(7.9) | 14(15.7) | 14(15.7) | 11(12.4) | 4(4.5) | 24 (27) | 65(73) | 29 | 60 | 4 | 8 | 11 | 19 | 31 | 16 | 54 | 48(53.93) | 4(4.49) | 2(2.25) | ||||||||
| Total Ve+ 65(73) | 89 | 89 | Total Ve+ 54(60.67) | ||||||||||||||||||||||||||
+ve, Leishmania positive; −ve, Leishmania negative; NI, not identified; W, wet; D, dry; F, female; M, male.
Our investigation showed the readily maneuver of L. major parasites through expanding distribution in the broad ranges of geographical confines at the center of Iran (Yazd province). All examined patients had typical lesions in Abarkouh district and the serous was taken from fresh lesions (Fig.1). The grade numbering of amastigotes is illustrated according to the WHO instruction in Table 1. The overall ratio of L. major infections were mainly higher than L. tropica with low density (Mann–Whitney U test: p < 0.05) based on the asymmetric distribution and non parametric analysis of the samples.
Leishmania parasites identification with RFLP and sequencing of ITS-rDNA fragmentThe DNA was isolated directly from the fresh ulcers sampling from 89 suspected patients and screened to amplify ITS-rDNA genotype by the research members of our team (Table 1). Of 89 Leishmania lesions, 65 slides confirmed microscopically indicating 54 molecularly positive samples (54/89, 60.67%) by amplifying ITS-rDNA fragment in agarose gel 1.5%. The positive ITS-rDNA samples were digested by BsuRI (HaeIII) and sequenced to identify Leishmania parasites at their species-specific level. Two samples did not have DNA enough to digest by BsuRI (HaeIII) and/or sequence. Given that of 54 molecularly positive samples, 48/89 (53.93%) were identified as L. major, four as L. tropica (4.49%) and two were remained ambiguous after resequencing (2.25%) (Table1).
RFLP analysis was done in CLC DNA Workbench 5.2 (CLC bio A/S, Aarhus, Denmark), two fragments of 120 and 310 bp belong to L. major, four (30, 40, 50 and 310 bp) for L. tropica and L. turanica and three (50, 70 and 310 bp) for L. Infantum. The pattern of BsuRI (HaeIII) enzyme confirmed as DNA fragments’ digestion in agarose gel after electrophoresis (Fig. 3). In addition, the two sequence of ITS-rDNA PCR products were compared to the GenBank sequences in case of similarity and homology. All sequences of Leishmania species (approximately 480 bp, including primers) were positioned in two different groups at which 52 Leishmania sequences had significant homology to each other and found as L. major (n = 48, 53.93%), L. tropica (n = 4, 4.49%) and two were unidentified (n = 2, 2.25) (Table 1).
Molecular analyses exhibited 48 positive L. major in a total of 65 microscopically positive lesions. 48 samples of L. major had two common haplotypes for L. major using the ITS-rDNA gene fragment GenBank access nos. EF413078 (Yaz32) and EF413077 (Yaz19) along with a common haplotype of standard strain (IROS/NA/76/ROSSI-II) form GenBank for L. tropica, accession no. AJ000302 (Yaz11). Also, three novel haplotypes were identified for L. tropica which had high variation in their nucleotide sequences GenBank accession nos. MH488992 (Yaz58); MH488993 (Yaz61) and MH492002 (Yaz60) (Fig. 4). Besides, the transition event of nucleotide substitution occurred in several positions of open reading frame (ORFs).

Maximum parsimony tree of the haplotypes of the ITS1-5.8SrRNA gene fragment for the isolates of L. major and L. tropica in humans of Yazd province with those submitted in GenBank using MEGA 5.05 software (details were shown in Table 1). *, Unique haplotypes; ⬤, Common haplotypes of Leishmania species identified in Abarkouh district, Central Iran; the standard reference strains of the World Health Organization are shown in the parenthesis.
Phylogenic tree showed that all Leishmania species were placed in their own exacting clade (Fig. 4). The topology of tree showed that gene duplication was occurred subsequent to speciation resulting from in-paralog event in a way that L. tropica and L. killicki were positioned in a Taxon of monophyletic clade.
48 L. major had common haplotypes incriminated previously in sandflies, rodents and humans in central and other places of Iran GenBank accession nos. EF413078 (Yaz32); EF413077 (Yaz19). But we found these only among the population of sandflies in Abarkouh district.2,5,6
DiscussionThis investigation has been kept evidential proof about L. major distribution with notable emerging of L. tropica in patients of Yazd foci, Iran. Current workup turned out that the species L. major had low genetic variation with predominant and high circulation rate whilst the species L. tropica exhibited high genetic diversity with different novel haplotypes and constant phenotypic features among the patients of Abarkouh district. Previously, we found more diversity in L. major using the ITS-rDNA genotype isolated from parasites in Iran.2,6 Although, L. major is well-known as a causative agent of ZCL in Iran,2,5,8,22L. tropica (n = 4; 4.49%) was isolated from the patients as a non-native parasite in the region. In addition, L. major was also found with high density (n = 48; 53.93%) in Abarkouh district.
The smears isolated from the chronic lesions of suspected patients had low amastigotes located more on hands, arms and foot respectively and they scored as +1 to +2 under microscopic observation. Although prepared smears from chronic old lesions had low amastigotes number microscopically, they had no Leishmania positive results after PCR and molecular analyses. In fact after doing molecular experiments on 22 microscopically positive samples from low grading numbers of amastigotes (n = 15, +1 and n = 7, +2), only 11 (n = 11/22) samples were confirmed positive and the rest (n = 11) were negative that this may be resulted from chronic lesion or DNA degradation in extraction procedures (Table 1). Also, smears isolated from freshly acute lesions were scored from +1 to +6 which they all were positive after molecular experiments. Freshly or acute lesions were sampled more from faces and hands respectively.17
Isoenzyme analyses were previously used as a gold standard-method for characterizing Leishmania species particularly in the old world23 but requiring the great numbers of live parasites in the culture and consequently the risk of being infected with other agents and/or fastest growth of any conqueror microbe was probable. Today, there is no single widely accepted method to specify Leishmania species stoutly. Thus, a wealth of molecular techniques is conducted to incriminate Leishmania parasites in primary infections.8,24 recent reports on vector and reservoir hosts of ZCL showed that only L. major was detected in female P. papatasi (15.8%) and within the different species of rodents in the villages of Abarkouh district.5,25 Likewise, Spotin and colleagues have recently reported the different morphologies of L. major amastigotes’ shapes but no molecular diversity observed in ZCL area of Iran.26P. papatasi is a proven vector of ZCL even in Isfahan (central) and Golestan (North east) provinces of Iran.16,27 Although, no L. tropica was previously detected in vectors of Abarkouh district,5 there was a striking difference in the detection of Leishmania species from principal vectors of L. tropica (Phlebotomus sergenti) and from human’s lesion as mammalian host in the same region. Molecular identification of L. tropica from skin lesions has shown that L. tropica is present in a considerable region at the center of Iran, Abarkouh district, and for the first time in the areas formerly known only as L. major foci.19,28 Hence, it is likely that Anthroponotic CL (ACL) has been considerably underreported in this district. However, because of existence of canines, stray dogs, sheep dogs and particularly domestic dogs as main reservoir hosts of L. tropica, more humans and dogs need to be monitored before concluding whether they are principal reservoir hosts of L. tropica in central Iran (Yazd province).29
Furthermore, infected humans with L. tropica parasites have been reported from Isfahan (central)30 and Fars, southern Iran.13,14 In addition, two provinces of Isfahan and Fars are adjacent to Yazd province (sympatric areas) and must be considered in case of patients travelling, epidemiological distribution of reservoirs and vectors, biodiversity of Leishmania parasites and their potential as neglected emergence disease.
Nonetheless, two PCR amplicons of Leishmania parasites had a noticeable incongruity between their sequences and were not readable even after several resequencing. These two unreadable sequences were isolated from lesions of infected patients in two villages of Abarkouh (Nosrat Abad and Ardi) (Table 1). Two unreadable sequences could be arise from inadequacy of DNA, mixed infections with other ZCL and/or non-pathogenic parasites, or containing two loci that PCR primers have greater tendency or homology to one of the loci in this case.31 Anyhow, Oryan and colleagues (2013) have observed the high genetic variation of L. major relating to their different clinical manifestations of cutaneous leishmaniasis based on minicircle kDNA in Fars province, southern Iran.32 But some issues including the probable existence of other Leishmania species might have been failed to notice if the PCR primers demonstrated haplotype specificity.
To determine intra and inter-species relationships and even to identify separation between strains, applying miscellaneous (nucleus, e.g. ITS-rDNA and/or mitochondrial, e.g. Cyt b) genes with different degrees of diversity needs to be done on the basis of different metabolic enzyme through systematic multilocus sequence analysis (MLSA) through.33 MLST approaches need to be applied as a precious tool to determine similarities between Leishmania species34 and to screen more patients in different ZCL foci, of Iran.
Also we have already assessed the sensitivity of two methods of semi-nested for minicircle kinetoplast (kDNA) and nested PCR of ITS-rDNA to detect Leishmania parasites in Phlebotomus papatasi which revealed the high infection rates of L. major in Isfahan villages close to Yazd province.9 Apart from geographical habitats, the isolated L. tropica parasites in this study found to be diverged from their ancestors with three novel haplotypes. All haplotypes of L. tropica parasites were placed in a monophyletic cluster of a unique group and this can be resulted from more nucleotide discrepancies in a few copy numbers of ITS-rDNA (nuclear) genotype of orthologous loci for this species-specific phylogenetics. Also, Paralog mechanism (tandem duplication) of homologous recombination (HR) in extra chromosomal circles may have occurred between direct repeated sequences35,36 to expand their gene families. Accordingly, Leishmania parasites would be able to increase their gene copy number37 and therefore survive and adapt in a different conditions.
Recent studies have highlighted that aneuploidy appears as a pervasive belief in Leishmania37,38 and some eukaryotic pathogens like Leishmania develop their rapid adaptation to environment changes associated with aneuploidy as a new model of adaptation in parasites with asexual or unicellular reproduction.39 Changes in chromosome number may not necessarily be present in all individual cells. Leishmania parasites are generally considered as a diploid organism with their own manifestation of genome plasticity.40,41 Whereas, increasing in copy number of the Leishmania genes can be expected in response to the conditions of environmental changes,42 genetic variation of L. tropica is perceived as a constitutive feature and/or effective strategy in parasite–vector–host interplay to maintain parasites’ life cycles in their human hosts in the region of our study.
In addition, mixed infections of the same macrophage with different species of Leishmania parasites were shown to be experimentally possible.42 Concomitant infection of L. tropica and L. major has already isolated from an Iranian patient with mucosal leishmaniasis (ML) in Fars province (Southern Iran) next to Yazd province.15 The recent investigation showed that in P. sergenti (the vector of L. tropica), L. turanica promastigotes were present until the defection of blood meal remnants and also proved that L. turanica develops late-stage infections in P. papatasi43 the proven vector of L. major in Iran.
Maybe it is possible that had we investigated hard on sandflies as vectors or rodents as reservoir hosts, more elucidation of Leishmania species might have found as L. tropica or L. turanica but these, are outside the scope of this paper.
To confirm the precise identification of Leishmania species in unknown, concomitant or mixed infection in vectors or reservoir hosts, the DNA samples are needed to be cloned.44
To determine the distribution of all species of Leishmania parasites that infect humans particularly in areas where foci of two species overlap and also to clarify the role of reservoir hosts (rodents or dogs) in maintaining of Leishmania parasites as co-infectors, molecular characterization and phylogenetic analyzes may have a pivotal approach to identify species-specific parasites in their potential sandflies as vectors.24 Therefore, simultaneous identification of Leishmania parasites in their vectors and reservoirs provides a route for more different comparative molecular studies prior to ancient methods.
Conflicts of interestThe authors declare no conflicts of interest.
The fund of this work was supported by Pasteur Institute of Iran and the grant was awarded to Prof. Parviz Parvizi by the Pasteur Institute of Iran. The authors would like to thank Abarkouh Health Care authorities and the personnel of National Institute of Health Research (NIHR) in Yazd for their assistance and support to facilitate easier communication with indigenous people from rural areas of Abarkouh district during this investigation. We would like to express our deep sorrow over the passing away of Mehdi Baghban and thank him for his kind assistance with the field work and laboratory experiments.
