

Yersinia enterocolitica is a widespread Gram-negative bacterium that causes gastrointestinal disease and other clinical manifestations in humans. Potentially pathogenic Y. enterocolitica has been isolated in Brazil, from human, environmental, food, and animal sources. Herein we report a genome sequence of Y. enterocolitica subsp. palearctica strain YE 19, serotype O:3, biotype 4, sequence type 18, with virulence determinants isolated from human blood in Rio de Janeiro in 2005. The results corroborate other findings that this strain harbors a set of virulence determinants that could play a role in host pathoadaptation and may also justify the successful dissemination of bioserotype 4/O:3 in Brazil. The presence of strains harboring all of these virulence genes in Brazil is a potential threat to young children and immunocompromised individuals, for whom yersiniosis are a significant source of morbidity and mortality. The results of a genomic data analysis will help understand the virulence of Brazilian strains and provide data for Y. enterocolitica studies worldwide.
Yersinia enterocolitica is an enteric pathogen with a wide range of clinical and immunological manifestations, which depend on age and physical condition of the patient. Responsible for intestinal diseases, the most frequent occurrence in infants and children, including enterocolitis with an inflammatory diarrhea; however, in older children and young adults the symptoms include acute terminal ileitis and mesenteric lymphadenitis (mimicking appendicitis). On the other hand, in some cases, extra-intestinal manifestations have also been reported, including urinary tract and respiratory tract infection (empyema), osteoarticular infection (reactive arthritis), erythema nodosum, infected mycotic aneurysm, axillary abscesses, and endocarditis.1,2Y. enterocolitica is the major causative agent of yersiniosis worldwide and is typically transmitted via the fecal–oral route to humans and animals.3Y. enterocolitica can be classified by biochemical characteristics and serotyped into six biotypes (BT) (1A, 1B, 2, 3, 4, and 5).1 Strains that are non-pathogenic to humans and animals belong to BT1A, whereas highly pathogenic strains belong to BT1B; BT2–5 are low to moderate-pathogenic strains. The serotype O:3 biotype 4 comprises about 80–90% of human isolates in Germany and Europe, with rising global relevance.4
Little attention has been given to this microorganism in Brazil, where it is still poorly studied. Although Brazil has no official data about the incidence of this pathogen, sporadic cases have been published and some studies demonstrate the pathogenic potential of strains isolated from many sources, including humans, the environment, food, and animals. The bioserotype 4/O:3 is the main bioserotype isolated from human clinical cases.5,6
The aim of this brief communication was to analyze the virulence profile and announces the genome sequence of Y. enterocolitica subsp. palearctica, strain YE 19, serovar O:3, biotype 4 isolated in 2005 from blood of a hospitalized patient in Rio de Janeiro. It was deposited in the Collection of Listeria (CLIST) located at the Bacterial Zoonoses Laboratory (LABZOO/Oswaldo Cruz Institute/Oswaldo Cruz Foundation) under the accession number CLIST 4117. Sequence type 18 (ST18) was determined by multilocus sequence typing (MLST) and by use of the Yersinia MLST website. The ST18 comprises the main Y. enterocolitica serotype O:3 sequence type based on MLST database.
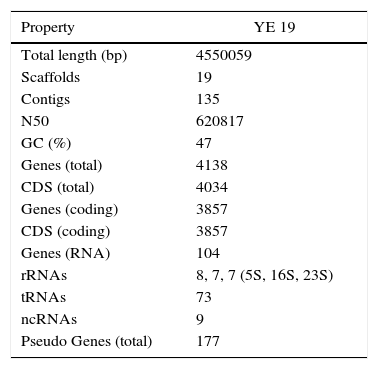
A next-generation sequencing (NGS) technique was used to analyze the virulome of YE 19 strain. Genomic DNA was extracted using DNeasy® Blood & Tissue Kit (Qiagen, Germany) and the DNA library was prepared with Nextera XT (Illumina, San Diego, CA). Sequencing was performed on an Illumina HiSeq 2500 platform (Illumina Inc., USA) generating 100bp paired-end reads. The sequence data were processed using Trimmomatic (v0.35) and paired-end reads were merged using FLASh (v1.2.11).7 De novo assembly was performed using SPAdes (v3.7.1).8 Ragout (v1.2)9 was utilized for reference assisted scaffolding of contigs against Y. enterocolitica subsp. palearctica Y11 genome (NC_017564.1). Sealer (v1.9.0)10 was used for gap filling and Pilon (v1.16)11 performed polishing. The resulting assembly was evaluated using REAPR (v1.0.18).12 The NCBI Prokaryotic Genome Annotation Pipeline (released 2013) annotated the final genome model comprising 19 scaffolds with a total length of 4,550,059bp and 47% GC. The summary of genomic statistics is reported in Table 1. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LYBL00000000. The version described in this paper is version LYBL01000000.
The presence of virulence factors was investigated using SRST2 program (v0.1.7) against the Virulence Factor Database (VFDB). The details of all virulence genes found are reported in supplementary file 1. Fig. 1 is built to show where each virulence gene is located on YE 19 genome.

A total of 103 virulence genes have been identified from the VFDB. These virulence genes are related to motility, the secretion system, toxins, the O antigen (lipopolysaccharide – LPS), hemin uptake, invasion, and adherence.
We detected virulence determinants of motility controlled by flhE, fliN fliL, flgA, fliD, fliC, fliP, flgD, flhA, fliS, flhB, flgK, fliF, flgB, flgJ, flgC, flgL, flgN, flgM, flhC, fliE, fliO, fliH, flgE, fliB, fliA, fliK, fliI, flgF, flgI, fliG, fliR, flgH, flhD, flgG, fliT, fliM, fliZ, and fliQ. These genes act in other processes, such as cellular invasion, biofilm formation, and the secretion system (the type III secretion/flagella biosynthesis apparatus).13,14 We found the cheA, cheB, cheD, cheR, cheW, cheY, cheZ genes, which act in the chemotaxis mechanism, by which bacteria efficiently and rapidly respond to changes in the chemical composition of their environment. This behavior modulates the direction of flagellar rotation.15 Finally, we found the motA and motB genes, which encode flagellar motor protein MotA and MotB, respectively, and build the flagellar-motor supramolecular complex stator.15
The yst1L, yst1J, yst1K, yst1F, yst1E, yst1G, yst1C and YE105_C0912 genes encode proteins that build the type II secretion system (T2SS). All Yersinia species, non-pathogenic as well as pathogenic, possess at least one of the two T2SS. The one T2SS that has so far been studied is termed Yts1, which is involved in dissemination and colonization of deeper tissues like liver and spleen in mouse infection experiments. Furthermore, the Yts1O gene encodes the Yts1O protein, which is a putative prepilin-like peptidase.16
YE105_C0309, YE105_C0310, YE105_C0312, YE105_C0313, YE105_C0314, YE105_C0323, YE105_C0324, YE105_C0325, YE105_C0326, YE105_C0327, YE105_C0328, YE105_C0332, YE105_C0333, YE105_C0334, YE105_C0335, YE105_C0336, YE105_C0338, YE105_C0339, YE105_C0340, YE105_C0341 are a set of genes that received the name of the strain from where they were recognized for the first time. These genes encode proteins that build the Ysc type III secretion system (pYV-encoded T3SS/TTSS). The T3SS is the mechanism by which pathogenic Yersinia injects proteins directly into the eukaryotic cell cytosol.3Y. enterocolitica has three different type III secretion systems, Ysc (pYV-encoded), Ysa (exclusive to biovar 1B), and the flagellar.2,3,13 Surprisingly, the genes found on YE 19 genome were related to the proteins (YscC, R, and U) that built the Ysc T3SS17 (Supplementary file 1). These findings reinforce the hypothesis that this strain already had the pYV and, somehow, these genes could be integrated into the chromosome. Meanwhile, further studies are needed to confirm this possibility.
We found the Yersinia stable toxin ystA gene, which encodes the production of a heat-stable enterotoxin that causes diarrhea.2
The presence of YE105_C0173, YE105_C1175, YE105_C1176, YE105_C1177, YE105_C1178, YE105_C1179, YE105_C1180, YE105_C1181, YE105_C1182 genes related to the O antigen were found. The O antigen or O polysaccharide chain is associated with antigenic properties and is involved in invasion and colonization processes. In addition, the O antigen is required for proper functionality or expression of other outer membrane virulence factors.2
We found the hemP, hemR, hemS, hemT, hemU and hmuV genes associated with hemin uptake. This set of genes permits the strain to use the heme-containing compounds as a source of iron.18 However, these genes are not the same genes found on yersiniabactin.2
The common virulence genes inv (invasive gene) and ail (attachment invasion locus) were present. They encode an invasin, which directly initiates cell penetration, and an outer membrane protein called Ail, which encodes a factor that also promotes the invasion of epithelial cells, respectively.1 The psaA (myfA) gene is related to adhesion to the host cell and its pH 6 antigen homolog in Y. pseudotuberculosis and Y. pestis.19 The myfB, myfC, myfE and myfF genes are encoded by the myf operon, known as the mucoid Yersinia factor. These genes encode proteins that constitute a fibrillar structure that acts during adhesion.2 The Ypla gene encodes phospholipase A that stimulates the acute inflammatory response of the host and is required for survival of Y. enterocolitica in Peyer's patches.3 Finally, the pla gene, which has been described in the Y. enterocolitica W22703 strain,20 encodes a peptidase A26 omptin protein that has proteolytic and adhesion functions.19 A Blast search on Genbank for this pla gene showed only 30% nucleotide identity with the pla gene from Y. pestis (strain I-3190 plasmid pPst); thus, further studies are needed to determine the relationship between these two genes.
These results corroborate the findings of Batzilla et al. and suggest that this strain harbors a set of virulence determinants that may play role in host pathoadaptation. This could also justify the successful dissemination of bioserotype 4/O:3 in Brazil and worldwide. As the pYV virulence plasmid1 was not found, we cannot rule out the possibility that it might have been lost during storage. The presence of strains harboring all of these virulence genes in Brazil is a potential threat to young children and immunocompromised individuals, for whom yersiniosis can represent a significant source of morbidity and mortality.3 We hope this genomic data analysis helps understanding the virulence issue of Brazilian strains and contributes for Y. enterocolitica studies worldwide.
Conflicts of interestThe authors declare no conflicts of interest.
We thank Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support.
The following are the supplementary data to this article:
Yersinia MLST website (available from: http://pubmlst.org/yersinia/) developed by Keith Jolley and sited at the University of Oxford (Jolley & Maiden 2010, BMC Bioinformatics, 11:595) [accessed in 08.29.16].
The Virulence Factor database (VFDB) (available from: http://www.mgc.ac.cn/VFs/) [accessed in 08.29.16].

- Home
- All contents
- Publish your article
- About the journal
- Metrics
- Download PDF
- Bibliography
- Additional material