



Cerebral malaria is the most severe and rapidly fatal neurological complication of Plasmodium falciparum infection and responsible for more than two million deaths annually. The current therapy is inadequate in terms of reducing mortality or post-treatment symptoms such as neurological and cognitive deficits. The pathophysiology of cerebral malaria is quite complex and offers a variety of targets which remain to be exploited for better therapeutic outcome. The present review discusses on the pathophysiology of cerebral malaria with particular emphasis on scope and promises of curcumin as an adjunctive therapy to improve survival and overcome neurological deficits.
Parasitic diseases have grown to become a health burden as around 30% of the world's population experience parasitic infections.1 Among various parasitic infections, malaria is the most life-threatening disease and accounts for 225 million clinical cases and 781,000 deaths per year worldwide of which 91% are in the African region. Beside this background, it is reported that each year about 85% of deaths globally are in children under five years of age, mainly due to the immunological factors.2 A total of US$ 38–45 billion will be spent from 2006 to 2015 for the diagnosis and treatment of malaria, mainly in underdeveloped countries which are the most affected by this deadly disease.3 Cerebral malaria (CM) is the most severe and rapidly fatal neurological complication of Plasmodium falciparum infection and responsible for more than two million deaths annually in non-immune individuals characterized by impaired consciousness.4 This represents an enormous burden of disease, due to the high prevalence of infection. According to the World Health Organization (WHO), CM is defined as a clinical syndrome characterized by coma (inability to localize a painful stimulus) at least 1h after termination of a seizure or correction of hypoglycaemia, presence of asexual forms of P. falciparum malaria parasites on peripheral blood smears and exclusion of other causes of encephalopathy.5
Till now, no effective vaccine is available against malaria because of the antigenic variation and complexity of parasite biology. The current therapy for management of CM is based on parenteral administration of either quinine or artemisinin derivatives such as artemether and artesunate. However, the emergence of drug resistance of P. falciparum to anti-malarials poses a serious challenge to malaria control. In addition, there are several non-parasitic events that contribute to pathogenesis of CM. The current therapy is aimed at reducing parasite burden and is inadequate in terms of reducing mortality or post-treatment symptoms such as neurological and cognitive deficits. These facts necessitate novel interventions for better therapeutic outcome in CM. Therefore, there is an urgent need to develop new antimalarial drugs, drug combinations or alternative strategies for the management of CM. The present review discusses on the pathophysiology of CM with particular emphasis on the use of curcumin, a natural polyphenol, as an adjunctive therapy.
Clinical manifestations of cerebral malariaThe clinical hallmark of CM is impaired consciousness with coma, generalized convulsions and neurological sequelae as the most severe manifestations. The earliest manifestations are non-specific fever, rigours and/or chills, irritability, restlessness or psychotic behaviour, vomiting and cough. The main symptoms in children are severe anemia, metabolic acidosis, hypoglycemia and coma initially arousable which becomes unarousable later. These clinical features are most strongly associated with an increased risk of death in children admitted to hospital with severe malaria. Gastrointestinal symptoms are common in children. In adults CM, renal failure, severe jaundice, and adult respiratory distress syndrome are the main complications. Ten percent of the adult patients develop severe intravascular hemolysis of infected and uninfected RBCs leading to hemoglobinuria and anemia which further contributes to renal failure. Pregnant women are also vulnerable and develop anemia, hypoglycemia, coma and pulmonary edema. Shock is not a common feature of severe malaria and if present may be due to septicemia.6,7
In children, coma develops suddenly followed by seizures in 80% of the cases following 1–3 days of fever but sometimes just after few hours. In contrast in adults, coma develops gradually and is occasionally associated with seizures. Clinical seizures are associated with rise in intracranial pressure. Status epilepticus is common in children and rare in adults. These patients often have deviated eyes, irregular breathing pattern, and show excessive salivation. On neurological examination, signs of meningism are uncommon but neck stiffness can be seen occasionally.8
Brain swelling, high intracranial pressure, changes in ocular movements, retinal changes (hemorrhages, peripheral and macular whitening, vessel discoloration and/or papilledema) and brainstem signs (abnormalities in posture, pupil size and reaction, ocular movements or abnormal respiratory patterns) are commonly observed. Various forms of abnormal posturing can occur in association with hypoglycemia. It can be either of decorticate or decerebrate pattern and often indicates raised intracranial pressure. Bruxism with grinding of teeth, pout reflex or brisk jaw jerk is common in cases with deep coma. Cranial nerve involvement is sometimes observed in adults but is rare in children.8,9
Clinical management of cerebral malariaManagement of severe malaria comprises of four main areas: clinical assessment of patient, specific antimalarial treatment, adjunctive therapy, and supportive care.
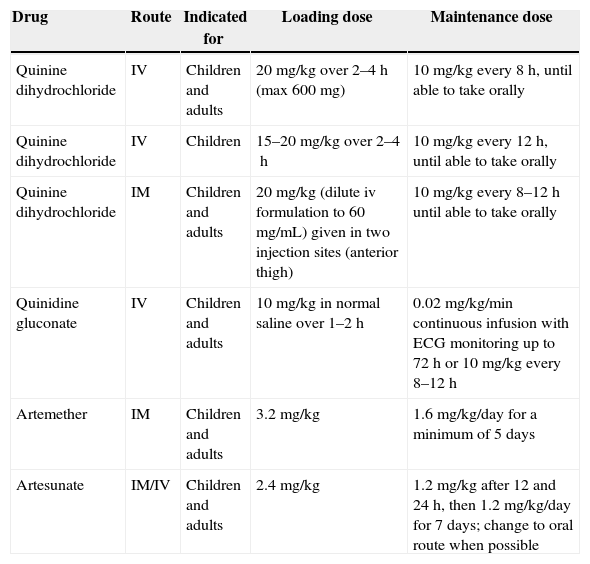
An open airway must be maintained in unconscious patients and breathing and circulation should be assessed. The patients should be weighed so that treatment can be done accordingly. Blood glucose, hematocrit/hemoglobin, parasitemia and renal functions must be assessed. The coma can be assessed using Glasgow scale in adults and Blantyre scale in children. Cerebrospinal fluid analysis shall be carried out to exclude bacterial meningitis as it may co-exist in endemic areas. Specific antimalarial treatment is described in Table 1. In attempt to reduce high mortality rate in severe malaria various adjunctive treatments have been suggested and are summarized in Table 2.
Treatment of cerebral malaria.
| Drug | Route | Indicated for | Loading dose | Maintenance dose |
|---|---|---|---|---|
| Quinine dihydrochloride | IV | Children and adults | 20mg/kg over 2–4h (max 600mg) | 10mg/kg every 8h, until able to take orally |
| Quinine dihydrochloride | IV | Children | 15–20mg/kg over 2–4h | 10mg/kg every 12h, until able to take orally |
| Quinine dihydrochloride | IM | Children and adults | 20mg/kg (dilute iv formulation to 60mg/mL) given in two injection sites (anterior thigh) | 10mg/kg every 8–12h until able to take orally |
| Quinidine gluconate | IV | Children and adults | 10mg/kg in normal saline over 1–2h | 0.02mg/kg/min continuous infusion with ECG monitoring up to 72h or 10mg/kg every 8–12h |
| Artemether | IM | Children and adults | 3.2mg/kg | 1.6mg/kg/day for a minimum of 5 days |
| Artesunate | IM/IV | Children and adults | 2.4mg/kg | 1.2mg/kg after 12 and 24h, then 1.2mg/kg/day for 7 days; change to oral route when possible |
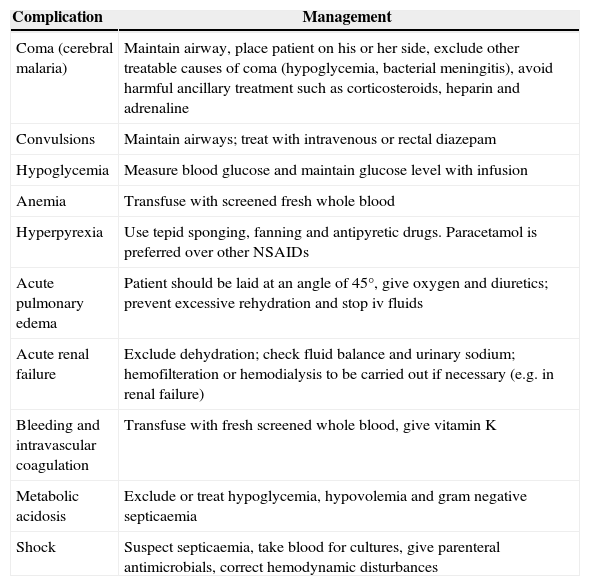
Clinical management of severe manifestations and complications of P. falciparum malaria.5
| Complication | Management |
|---|---|
| Coma (cerebral malaria) | Maintain airway, place patient on his or her side, exclude other treatable causes of coma (hypoglycemia, bacterial meningitis), avoid harmful ancillary treatment such as corticosteroids, heparin and adrenaline |
| Convulsions | Maintain airways; treat with intravenous or rectal diazepam |
| Hypoglycemia | Measure blood glucose and maintain glucose level with infusion |
| Anemia | Transfuse with screened fresh whole blood |
| Hyperpyrexia | Use tepid sponging, fanning and antipyretic drugs. Paracetamol is preferred over other NSAIDs |
| Acute pulmonary edema | Patient should be laid at an angle of 45°, give oxygen and diuretics; prevent excessive rehydration and stop iv fluids |
| Acute renal failure | Exclude dehydration; check fluid balance and urinary sodium; hemofilteration or hemodialysis to be carried out if necessary (e.g. in renal failure) |
| Bleeding and intravascular coagulation | Transfuse with fresh screened whole blood, give vitamin K |
| Metabolic acidosis | Exclude or treat hypoglycemia, hypovolemia and gram negative septicaemia |
| Shock | Suspect septicaemia, take blood for cultures, give parenteral antimicrobials, correct hemodynamic disturbances |
Patients with severe malaria require intensive nursing care. Clinical observations such as coma and other vital signs should be monitored regularly. Blood glucose should be monitored every 4h. Fluid requirement should be assessed individually. Patients with intravascular coagulation should be given fresh whole blood transfusion and vitamin K. Any concomitant infections should be treated with antibiotics based on culture and sensitivity results.
Pathophysiology of cerebral malariaMalaria is a protozoal disease of humans and its neurological complication, CM is a multisystem multi-organ dysfunction which is arguably one of the most common non-traumatic encephalopathies in the world.10 The pathogenesis of CM is still not fully understood because the animal models do not recapitulate human disease completely. Thereby, in vivo and in vitro models are useful for hypothesis generation but proof of causation in clinical pathogenesis, naturally, must be investigated in human patients. The combination of both parasite and host factors is involved in the pathogenesis of CM.
The pathogenesis of CM results from the adherence and sequestration of parasitized red blood cells (PRBCs), immune cells and platelets to vascular endothelial cells (ECs) lining the small blood vessels of the brain leading to their blockade.11 This is further accompanied by over production of type-1 pro-inflammatory cytokines followed by upregulation of endothelial adhesion molecules (Fig. 1).12 It leads to micro- and ring-hemorrhages and necrosis of the surrounding tissues and brain edema resulting in a significant compression of cerebral arteries that could ultimately represent the cause of death.13 The symptoms of CM range from confusion or stupor to obtundation, convulsions and deep coma with long-term neurological deficits such as cortical blindness.14
 breakdown during cerebral malaria pathogenesis. The diagram shows the events and the possible mechanisms that play a vital role in CM. The mature forms (trophozoites and schizonts) of parasitized red blood cells (PRBCs), host leukocytes and platelet-fibrin thrombi adhere to the cerebral endothelial cells and sequester in large numbers in the brain. This cytoadherence, combined with other events such as rosetting, auto-agglutination, clumping and decreased RBCs and PRBCs deformability causes altered blood flow leading to impaired tissue perfusion and hypoxia. Further, sequestered parasites produce local toxins which lead to recruitment of leukocytes and platelets followed by the release of inflammatory cytokines (IL-1, IL-6, TNF, LT, and NO) and microparticles. These mediators lead to endothelial cells activation and apoptosis, BBB breakdown, increased junctional permeability, followed by secondary neuropathological events that can lead to cerebral edema or coma. Sequestration of PRBCs within microvasculature increases cerebral volume, which together with increased cerebral blood flow from seizures, anemia and hyperthermia and altered BBB function lead to cerebral edema and raised intracranial pressure. This may result in death or neuronal damage with consequent neurocognitive sequelae in the survivors.")
The blood–brain barrier (BBB) breakdown during cerebral malaria pathogenesis. The diagram shows the events and the possible mechanisms that play a vital role in CM. The mature forms (trophozoites and schizonts) of parasitized red blood cells (PRBCs), host leukocytes and platelet-fibrin thrombi adhere to the cerebral endothelial cells and sequester in large numbers in the brain. This cytoadherence, combined with other events such as rosetting, auto-agglutination, clumping and decreased RBCs and PRBCs deformability causes altered blood flow leading to impaired tissue perfusion and hypoxia. Further, sequestered parasites produce local toxins which lead to recruitment of leukocytes and platelets followed by the release of inflammatory cytokines (IL-1, IL-6, TNF, LT, and NO) and microparticles. These mediators lead to endothelial cells activation and apoptosis, BBB breakdown, increased junctional permeability, followed by secondary neuropathological events that can lead to cerebral edema or coma. Sequestration of PRBCs within microvasculature increases cerebral volume, which together with increased cerebral blood flow from seizures, anemia and hyperthermia and altered BBB function lead to cerebral edema and raised intracranial pressure. This may result in death or neuronal damage with consequent neurocognitive sequelae in the survivors.
Cytoadherence is defined as adherence of PRBCs to the vascular endothelium, mediated by Plasmodium derived proteins on the surface of PRBCs and modified erythrocyte cell wall proteins and ligands on the ECs.15 The best described adhesin protein is parasite protein PfEMP-1 (P. falciparum erythrocyte membrane protein-1) which is a large transmembrane protein (200–350kDa) comprising several external Duffy-binding-like (DBL) domains. It is encoded by a family of more than 150 highly variable (“var”) genes.16 These are knob-like electron dense structures protruding from the PRBCs surface. The role of the knobs in cytoadherence was confirmed when knobless variant of P. falciparum were shown to lack adherence to host cells in vitro as well as in vivo.17 Surface potential spectroscopy studies of knobs revealed that they were positively charged and hence better electrostatic interaction with negatively charged endothelial plasma membranes and receptors.18 These high molecular (200–350kDa) transmembrane proteins are encoded PfEMP-1 mediated cytoadhesion begins at approximately 12h of parasite development and is highly effective in the second half of P. falciparum life cycle. As a result, late stage of parasite (trophozoites and schizonts) is rarely seen in peripheral blood because of their sequestration in various organs.19 The large number of variants of PfEMP-1 results in potential of infected RBCs to bind to many different host receptors expressed on vascular ECs in peripheral microvascular beds.20 There are several cell adhesion receptors that have been characterized including ICAM-1 (intercellular adhesion molecule-1), VCAM-1 (vascular cell adhesion molecule), CD36, CD31, thrombospondin, E-selectin, hyaluronic acid and chondroitin sulfate A (CSA).8 Cytoadherence prevents malarial parasites from being recognized and cleared off the circulation by the spleen.21
CD36 seems to be constitutionally expressed on wide range of vascular beds but remarkably absent in brain vessels and thus its expression is not related qualitatively or quantitatively to disease severity.22 This receptor is present in the microvasculature and its expression is not affected by inflammatory cytokines like tumour necrosis factor-α (TNF-α).23 However, platelets expressing CD36 may serve as a stick bridge between PRBCs and endothelium which could be important in cerebral vessels lacking CD36 CSA is the main receptor in the placenta which in addition to CD36, are able to support firm adhesion under flow conditions.24 The E-selectin mediates initial tethering to the ECs and rolling of leukocytes. Plasma concentration of soluble E-selectin is raised in P. falciparum malaria.25
Main receptor for the cytoadherence is ICAM-1, which has been implicated in the pathogenesis of CM. It is a membrane glycoprotein expressed on lymphocytes, macrophages and vascular endothelium. Its primary role is to mediate cellular adhesion within the immune system via the leucocyte integrins LFA-1 and MAC-1.26 Expression of ICAM-1 on human vascular endothelium can be upregulated by the pro-inflammatory cytokines, TNF-α, Interferon-γ (IFN-γ) and Interleukin 1-α (IL 1-α).27 Regulation of ICAM-1 was shown to involve the endothelial nuclear factor kappa beta (NFκB) system and may involve reactive oxygen species (ROS).28 NFκB is a transcription factor located within cytoplasm and responsible for regulation of cytokine production. Normally NFκB stays inactivated by an inhibitory protein IκB. Once activated, NFκB enters the nucleus and increases the transcription of different inflammatory mediators such as cytokines and chemokines (TNF-α, IL, etc.) thereby causing neuroinflammation. Several molecules are capable of activating NFκB including TNF-α29 Monoclonal antibodies have been developed against ICAM-1 to improve microcirculatory flow in ex vivo models of malaria sequestration but have not been investigated in humans.30 Recently, it has been reported that cytoadherence, a potential pathogenic property of P. falciparum infected red blood cells, continues long after the parasite has been killed.31
SequestrationSequestration of PRBCs containing mature forms of the parasite (trophozoites and meronts) in the cerebral microvessels has been found on post-mortem examination of the CM patients.32 The pathogenesis of fatal CM is associated with sequestration of red blood cells. It has been confirmed that severe malaria pathology is related to sequestered parasite biomass.33 Sequestration occurs principally during the second half of the intra-erythrocytic asexual growth phase of the parasite, following the cytoadherence. The accumulation of mature PRBCs is observed in almost all organs, but particularly in the brain viz. cerebrum, cerebellum and medulla oblongata.11,32 This in turn, reduces the vascular lumen to create a mechanical obstruction for the erythrocytes transit. It is the sequestered parasites that cause considerable obstruction of blood flow, decreases tissue perfusion and removal of waste products like lactic acid and generates hypoxia.9,33
The relatively hypoxic venous beds provide optimal environment for the optimal growth and multiplication of parasite and prevents the PRBC from being destroyed by the spleen.34 Tissue hypoxia further causes hyperlactemia with an increased lactate pyruvate ratio and acidosis which are the strongest reproducible prognostic indicators in falciparum malaria, both in children and adults.35
Red cell deformability, rosetting and auto-agglutinationParasitized erythrocytes and non-parasitized erythrocytes (NPRBCs) sequestration in the cerebral microvasculature reduces the microvascular flow. In addition, with progressive maturation of parasites inside erythrocytes decreases their ability to deform which may contribute to the erythrocyte destruction and impair the microcirculatory flow.15 Decreased deformability of PRBCs is mainly due to change in the cytoskeleton, increased stiffness of the membrane and rigidity of growing intracellular parasite. Such PRBCs have more difficulty in passing through in vitro micropore filters as demonstrated for Plasmodium knowlesi malaria in monkeys.36 Furthermore, it has been observed that NPRBCs have to undergo considerable deformation as they squeeze through the sequestered microcirculation. It can be concluded that deformability of both PRBCs and NPRBCs is markedly reduced in severe malaria which is partly contributing to the pathogenesis of CM.37
The adherence of NPRBCs to PRBCs (rosetting) and PRBCs to PRBCs (agglutination) could further compromise microcirculatory flow that leads to impaired tissue perfusion, hypoxia and creates a toxic local environment.15 This phenomenon is more prevalent in patients with CM than in patients of uncomplicated malaria. The var genes seem to be responsible for rosetting and are sensitive to pH and heparin.38 Rosettes can be disrupted by antibodies against P. falciparum, glycosaminoglycans and sulfated glycoconjugates which is strain-specific.39,40 Rosette formation in vitro indicates the clogging of vascular lumen by interaction of NPRBCs and PRBCs with endothelium. However, PRBCs causes the cytoadherence but not all form rosettes.41 Interestingly, epidemiological studies have shown the association of rosette forming strains with the development of severe disease but most clinical studies have failed to show such an association.38,42 Red blood cells in thalassemic and sickle cell anemia patients have weak binding abilities forming small and weak rosettes. This decreased rosetting ability provides natural protection against development of CM in such patients.43 Thus, it can be concluded that a compromised microcirculation, with sequestration of parasitized erythrocytes due to reduced red cell deformability and the sticky forces related to rosetting and auto-agglutination, is central in the pathogenesis.
Inflammatory mediatorsP. falciparum infection causes an increase in blood concentration of both pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, IL-18 and anti-inflammatory Th2 cytokines IL-4 and IL-10. The balance between these inflammatory mediators is critical to parasite control. However, their role in pathogenesis of disease remains unclear.44 Studies have shown that glycosylphosphatidylinositol (GPI), a malarial toxin, binds to the pattern recognition receptors of the innate immune system and stimulates pro-inflammatory cytokine production.45 GPI stimulates the production of TNF-α and lymphotoxin which further cause upregulation of expression of cytoadherence receptors notably ICAM-1 and VCAM-1 on ECs. Thus, they promote sequestration of PRBCs and leukocytes in brain leading to coma. The biological effects of TNF-α during malaria are both protective and pathogenic.37,46 At low concentration, TNF-α kills parasite by causing cytokine release from macrophages. Higher concentration of TNF-α is associated with hypoglycemia, hyperparasitemia, anemia, pulmonary edema and death.47
It has also been found that TNF activation leads to the production of microparticles which have pro-coagulant and pro-inflammatory properties and this is specifically seen in patients suffering from CM and does not occur in uncomplicated malaria or severe anemia.48 TNF-α also induces the release of nitric oxide (NO) via inducible NO synthase (iNOS) which has been proposed as cause of coma due to interference with synaptic transmission.49
The effect of NO in malaria could be either beneficial or harmful depending upon the amount and site of production. Beneficial effect of NO is involved in host defence by killing intracellular organisms, maintaining vascular status and neurotransmission.9 Although, the direct killing effect of NO on the parasite was not observed, but oxidation products of NO were found to be toxic to the malarial parasite.50 Moreover, it was also shown that human monocytes stimulated with IFN-γ via the secretion of reactive nitrogen intermediates had inhibitory action on parasite growth in vitro.51 Measurement of reactive nitrogen intermediates in various biological fluids such as plasma, cerebrospinal fluid and urine is very difficult since NO is short lived, unstable and reacts rapidly with several molecular targets. This observation correlates poorly with in vivo production of NO at microvascular level. NO can easily diffuse across blood–brain barrier (BBB) and reduces the level of consciousness, impair neuronal signalling at higher levels and causes oxidative damage of ECs. Furthermore, NO may damage RBCs and increase anemia. To counteract the damage caused by the radical, NO scavengers might represent an effective adjunct therapy. An alternative hypothesis states that NO production is limited and it is synthesized by scavenging molecules such as superoxides and plasma hemoglobin. Thus, enhancing NO bioavailability has been proposed as anti-malarial therapy.52 However, the role of NO in CM pathogenesis has not been elucidated clearly and conflicting observations have been reported.8
The cause of impaired consciousness is still unclear but it is likely to result from the in homogenous obstruction of the cerebral microcirculation by sequestered parasitized erythrocytes causing dysoxia and results in lactate production by the brain.53 Apart from NO mediated loss of consciousness various host and parasite derived factors may be involved in pathogenesis of coma. Further, autopsy studies indicate the axonal dysfunction which may arise due to accumulation of β-amyloid precursor protein. This could in turn contribute to the reversible neurological dysfunction in CM.54 However, the cause of coma in CM and the role of dysoxia and axonal dysfunction still remains unclear.
Reactive oxygen speciesSevere malaria patients are associated with an increased production of ROS by the phagocytic cells which in turn are responsible for the tissue damage. However, it was suggested that ROS plays a role in host defense mechanism against the parasite.55 Thus, likewise NO, ROS too can have both beneficial and pathological roles.
Oxidative stress (OS) plays a vital role in malaria parasite-infected erythrocytes and at the same time OS represents a most promising rationale for designing the anti-malarial chemotherapy. This has been indicated by the glucose-6-phosphate dehydrogenase deficiency which makes the malarial parasites highly susceptible to alterations in the redox equilibrium. This in turn contributes to disease manifestation including sequestration, cerebral pathology, anemia and respiratory distress.56 However, the redox interactions between the malarial parasite and that of their host are complex. Due to high metabolic rate of the rapidly growing and multiplying parasite, detoxification of ROS becomes a key challenge for infected erythrocytes. Central to the generation of toxic redox-active-by-products is the degradation of host hemoglobin by the Plasmodium. Hemoglobin represents the major source of aminoacids for Plasmodium, but its degradation in an acidic food vacuole results in the production of toxic free heme (ferri/ferroprotoporphyrin IX; FP) and ROS which in turn confers oxidative insult on the host cell. Hemozoin or malaria pigment is a crystalline form of free heme. Even if only a small amount (e.g. 0.5%) of the free heme escapes the neutralization processes, it could cause OS to host proteins and membranes, inhibit parasite enzymes and hemolysis.57 In addition, the production of ROS by the host immune system also contributes to the overall oxidative burden of the infected erythrocyte. In order to maintain the redox status, malarial parasite possesses an efficient enzymatic antioxidant defense system including glutathione- and thioredoxin-dependent proteins as well as superoxide dismutase.56 In this regard, targeting the parasite antioxidant defense system might be useful for designing antiparasitic medication.
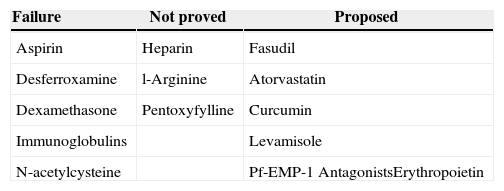
Indeed, a number of potential new drugs currently in clinical use, especially the artemisinins appear to act by alkylation of heme or protein to form a cytotoxic carbon-centred radical intermediate that further interfere with the redox metabolism of malarial parasite.58 It seems likely that the lethal effects of endoperoxide antimalarial are due to targeted damage of particular enzymes or membrane proteins by free radical species rather than non-specific damage caused by freely diffusing ROS.59 Some antioxidants adjunctive therapy have been evaluated in humans and mice (Table 3).
Increased permeability and evidence of endothelial damageThe BBB is a highly specialized structural and functional interface between the intraerythrocytic stages of P. falciparum and the human host.60 BBB maintains functional integrity with the support of pericytes, microglial processes of neurons and specialized perivascular macrophages. Perivascular cells are involved in maintaining the normal neuronal function of the CNS and provide immunity.61 There is growing evidence that combination of parasite-induced sequestration of PRBCs and NPRBCs, cytokine response, host environment, activation of ECs and hemostasis changes BBB function and its permeability. This has been implicated in the onset of a clinically complex syndrome of coma with reversible encephalopathy, associated with high mortality rate and increasingly recognized as long-term neurological sequelae in survivors.8,62
Pathological changes to BBB dysfunction may be broadly divided into systemic effects of severe disease such as acidosis or hypoxia due to anemia and local factors include adherence of PRBCs, host leukocytes and platelets to the ECs, rosetting, sequestration, microvascular obstruction, release of cytokines and parasite toxins. These cascade of events cause direct cellular damage and may initiate signalling within ECs that could influence junctional protein expression, and also directly activate or damage peri- and paravascular cells such as microglia, astrocytes and neurons and parenchymal effector cells.14 It is believed that cerebral microvascular engorged by cytoadherent parasitized erythrocytes results in a cascade of intracellular signalling events that disrupts the cytoskeleton-cell junction structure and increases the junctional permeability of the BBB and suppresses activation of dendritic cell and macrophages.12 Thus, dysfunction of BBB initiates the progression of secondary processes of neuropathological injury.
Even though P. falciparum disturbs the BBB integrity, it never enters the brain parenchyma and yet it can cause severe neurological dysfunction and death.14,61 Focal disruptions at sites of PRBCs sequestration along the ECs of BBB could result in the exposure of sensitive perivascular neuronal cells to plasma proteins and further leads to localized hypoxia and hemorrhaging of surrounding brain parenchyma.12,14 Apart from systemic effects, increased concentration of cytokines and parasite toxins have also been shown to contribute to reduced consciousness and seizure activity. Thus causing direct damage to BBB.63
Studies have shown that PRBCs adhesion to ECs induces cell apoptosis via over expression of cytoadherent molecules, expression of iNOS and OS.64 Further, in vivo studies in mice model of CM have implicated cytotoxic effector CD8+T lymphocytes in development of murine CM pathogenesis by direct cytotoxicity against ECs.65 Thus, apoptosis and cytotoxicity may lead to disruption of BBB and endothelial damage.
In patients, CM-associated brain damage can be studied only on post-mortem specimens and is the end-point of a fatal syndrome. Post-mortem analysis of infected brain tissue from severe P. falciparum cases has shown ECs activation, vascular congestion, varying degree of cerebral edema, leakage of plasma into brain interstitium, upregulation of ICAM-1, petechial hemorrhage particularly of white matter and ring hemorrhages in white matter.9,15
The leakage of plasma into CSF and consequent elevation of its protein concentration due to breakdown of BBB has been demonstrated by parenteral administration of radioactive albumin and isothioacyanite in animal models.66
BBB disruption can contribute to severe increased intracranial hypertension which is associated with neurological sequelae or death. However, the most likely cause of raised intracranial hypertension includes increased cerebral blood volume due to sequestration and increased blood flow from seizures, hyperthermia and anemia. Mannitol is an effective osmotic diuretic in reducing the burden of brain edema and intracranial pressure in children with mild degree of intracranial hypertension. It may decrease the mortality or neurological deficits in surviving patients of CM.67
Neurocognitive deficitsThough most of the patients suffering from CM make a full recovery, survivors have been found to be associated with neurological sequelae.9 CM sequelae seem to be of multifactorial origin, although their cause is largely unknown. These impairments are rarely observed in adults (<1%) but are more common in children (approx∼12%) particularly in sub-Saharan Africa.68 The symptoms include hemiplegia or quadriparesis, cortical blindness, deafness, ataxia, severe hypotension and aphasia.69 The sequelae reported include protracted seizures, prolonged and deep coma, hypoglycemia and sometimes severe anemia.9 Some of these symptoms resolve over a period of 1–6 months in 50% of the children. However, 25% of children with successfully treated CM are left with residual cognitive and neurological deficits such as memory disturbances, speech and language difficulties, disorders of attention, seizures, visuo-spatial and motor deficits.70 These deficits have been determined to persist after cessation of therapy with follow-up periods extending to nine years after infection.71 These cognitive deficits occur as a result of complicated host inflammatory response in conjunction with vascular impairment, causing abnormalities in neuronal cells, which adversely affect cognitive function.
Prolonged febrile seizures not precipitated by malaria may lead to damage in the hippocampus associated with later episodic memory impairments which may be underreported.9,15,72 Further decreased in blood flow and delivery of oxygen may lead to brain damage.15 The other reasons include generation of excitotoxins, ROS, and toxins produced by the parasite.73
To date, there is no therapy targeting the cognitive and motor deficits seen after successful antimalarial therapy in experimental CM. Hence, the role of immunomodulator needs to be assessed as an adjunctive therapy in CM.
Severe Plasmodium vivax infections and neurological complicationsP. vivax infections considered as a benign disease for a long time is the major cause of morbidity in Asia, Central and South America and in the horn of Africa.74 It is responsible for more than 400 million infections each year thus representing most widespread malarial species.75 In 2010, 306,908 cases of malaria were recorded in Brazil, 85% of which were caused by P. vivax.76P. vivax infections are characterized by relapse of malaria due to persistent latent forms in liver called as hypnozoites. In most of endemic areas, P. vivax infections co-exist with P. falciparum infections. However, P. vivax infections are more difficult to control because of their tendency to relapse and is a major cause of malaria in young children.74 Severe P. vivax infections are uncommon but have been reported at regular intervals. The number of vivax infections particularly those with serious clinical complications has been increasing. The most frequent complications are severe anemia, acute respiratory distress syndrome (ARDS), thrombocytopenia, renal and hepatic dysfunction, coma, CM and death.77P. vivax infections have been rarely associated with CM and to date less than 50 cases of central nervous system P. vivax infections have been reported.78 Severe pulmonary involvement is frequent in P. falciparum infection but rare in vivax malaria. Among the few cases reported involving vivax, the pulmonary complications appear from six to eight days after the initiation of antimalarial treatment. This is due to exacerbation of inflammatory response.79 However, in contrast to P. falciparum associated respiratory distress, the lungs are the sole organ system involved in vivax malaria. It is associated with acute lung edema and neutrophil accumulation in interalveolar space with or without sequestration of P. vivax infected RBCs.77,80
The mechanism of ARDS in vivax malaria is less well studied compared to falciparum infections. The level of pro-inflammatory mediators is elevated in vivax infections thus signifying the role of inflammatory dysregulation. There is a cytokine related increase in alveolar permeability and altered fluid clearance.81 In vivax infections, anemia persists even after the parasite clearance. The severe anemia in vivax infections does not result from destruction of infected RBCs alone. Studies have shown that for every infected RBC destroyed in vivax malaria, 32 uninfected RBCs are removed from circulation compared to eight RBCs in falciparum malaria.82
Immunopathogenesis of vivax malaria is poorly understood. However, it was found that Brazilian patients with severe vivax malaria had elevated levels of inflammatory cytokines TNF-α and IFN-γ, increased IFN-γ/IL-10 ratio and superoxide dismutase (SOD-I).83 The level of TNF-α and other inflammatory cytokines are higher during P. vivax infections compared to P. falciparum infections with same parasite load. Plasma concentration of microparticles was found to be elevated in individual with P. vivax infections.84 In contrast to P. falciparum, P. vivax has low parasite biomass and is considered unable to cytoadhere or sequester. However, recently P. vivax infected RBCs have been shown to cytoadhere to ECs via chondroitin sulfate-A and ICAM-1 although at 10-fold lesser frequency than P. falciparum.85 This could be reason for lower incidence of coma in vivax malaria and coma is perhaps of systemic metabolic origin.86 The degree of endothelial activation is greater in vivax than falciparum malaria. Rosetting in P. vivax has not been well documented and has not been associated with disease severity. The difficulty of growing P. vivax in vitro and absence of specific and reliable biomarkers have lead to little knowledge about its pathogenesis and severe cases. In areas where both multidrug resistant P. falciparum and P. vivax are prevalent, infants and young children are at greater risk of acquiring P. vivax infection than P. falciparum infection.87 Thus, an immunomodulator may have a potential role in severe vivax infections.
Need of adjunctive therapy in cerebral malariaApart from P. falciparum induced cerebral injuries, CM is also associated with non-parasite events including OS and inflammatory cascade which is responsible for mortality in spite of antimalarial treatment during the acute phase.88 CM is associated with elevated pro-inflammatory cytokines in serum and brain tissues, as well as leukocyte recruitment to sites of parasite sequestration in the cerebral microvasculature. The harmful, deregulated immune response leading to CM is mainly of the Th1 type, with overproduction of Interferon-γ (IFN-γ) and TNF-α.89 These inflammatory processes promote pathological changes in the brain that contribute to CM, such as endothelial damage and neuronal dysfunction. In spite of treatment with current antimalarials, CM is associated with mortality in 20–25% of cases and those surviving have been associated with neurological and cognitive deficits.71 OS has been found as a contributing factor to persistent cognitive deficits.90 ROS are generated in malaria as a result of host immune response and may oxidize vital cellular components such as lipids, proteins, and DNA. Oxidative damage to membranes of parasitized and uninfected red blood cells may contribute to sequestration of red blood cells in microvascular cells.91 The current therapy utilizing antimalarials controls parasitic events alone and do not treat the inflammatory cascade. The control of these deleterious processes is probably one of the most challenging goals to reach for a full rate recovery suggesting that adjunctive therapies are urgently needed with the aim of providing a new therapeutic scheme for CM using a combination of a rapid antimalarial and immunomodulators. Also, the reversal of BBB leakage or its downstream effects presents a potential opportunity for neuroprotective therapy for patients with CM. Apart from killing the parasite effectively, there is a need to improve survival rate and overcome cognitive deficits in CM patients and therefore reduce the likelihood of permanent neurological disability or death. Thus, the objectives of the adjunctive therapy are mainly to decrease the mortality, shorten the coma and reduce the rate and the severity of neurological sequelae. The first adjunctive intervention was using anti-TNF antibody in the murine model and since then more than 48 adjunctive interventions have been evaluated showing 92% success rate in ameliorating or preventing CM.92 The adjunctive therapy may involve the identification of immune modifying compounds such as curcumin, with ability to downregulate excessive pro-inflammatory response, reducing the expression of the adhesion molecules and subsequent sequestration of PRBCs in cerebral endothelium, thus improving the survival of CM patients. In contrast to many adjunctive therapies which have been investigated, curcumin has an added advantage of possessing antimalarial efficacy of its own. An important point to be considered for administration of adjunctive therapy using an immunomodulator is that it should be administered together with antimalarial drugs rather than alone in order to delay immunopathology and allow a longer time frame for antimalarial to work.4 We are working on intranasal delivery of curcumin loaded nanoemulsions in combination with artemether for management of CM.93
Curcumin and cerebral malariaCurcumin, a small molecular weight polyphenolic compound isolated from the rhizomes of Turmeric (Curcuma longa L.) and related species (family Zingiberaceae), has been used traditionally as a spice. The major curcuminoids present in turmeric are Curcumin (Curcumin I), Demethoxy curcumin (Curcumin II) and Bisdemethoxy curcumin (Curcumin III) (Fig. 2). Commercial curcumin contains curcumin I (77%), curcumin II (17%) and curcumin III (3%).94

Curcumin is from natural sources of long-term use, and as such, no resistance is known to curcumin that is present in a dietary supplement. It is tolerated at very high doses, and as much as 8g/day has been given for three months to cancer patients on trial without toxic side effects.95 A large number of in vitro and in vivo studies in both animals and humans have indicated that curcumin exhibits promising pharmacological activities including antioxidant, anti-inflammatory, anti-angiogenic, anti-spasmodic, anti-microbial and anti-plasmodial activities. Accumulating evidence suggests that curcumin has a diverse range of molecular targets, supporting the concept that it acts upon numerous biochemical and molecular cascades (Fig. 3).

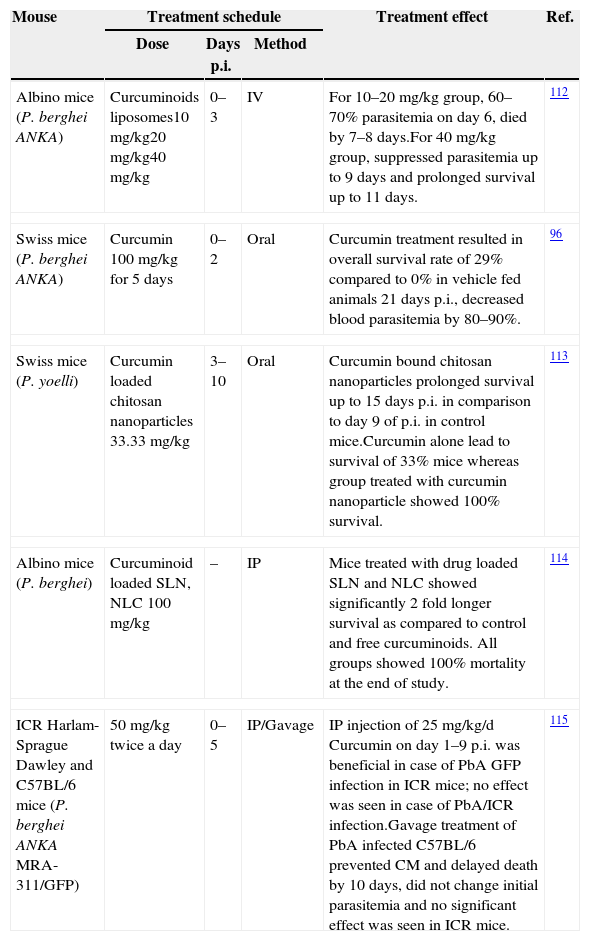
In the context of malaria, curcumin has been shown to possess moderate antimalarial activity with IC50 value of 5–18μM.96,97 Further, curcumin analogues were synthesized having much more potent antimalarial activity with IC50 value of 400nM. With the use of the CM model in mice, it has been shown that curcumin have potent activity against Plasmodium berghei and it is able to prevent CM and delay death of animals by about 10 days.4,98 In addition to having a direct killing effect as an antimalarial, curcumin is also able to prime the immune system against P. berghei.98 Curcumin possesses both anti-oxidant and pro-oxidant activity. Curcumin has shown to enhance ROS generation in concentration range of 20–100μM.99 The efficacy of curcumin has been tested in different models of CM (Table 4).
Curcumin examined for possible effects on animal models of malaria.
| Mouse | Treatment schedule | Treatment effect | Ref. | ||
|---|---|---|---|---|---|
| Dose | Days p.i. | Method | |||
| Albino mice (P. berghei ANKA) | Curcuminoids liposomes10mg/kg20mg/kg40mg/kg | 0–3 | IV | For 10–20mg/kg group, 60–70% parasitemia on day 6, died by 7–8 days.For 40mg/kg group, suppressed parasitemia up to 9 days and prolonged survival up to 11 days. | 112 |
| Swiss mice (P. berghei ANKA) | Curcumin 100mg/kg for 5 days | 0–2 | Oral | Curcumin treatment resulted in overall survival rate of 29% compared to 0% in vehicle fed animals 21 days p.i., decreased blood parasitemia by 80–90%. | 96 |
| Swiss mice (P. yoelli) | Curcumin loaded chitosan nanoparticles 33.33mg/kg | 3–10 | Oral | Curcumin bound chitosan nanoparticles prolonged survival up to 15 days p.i. in comparison to day 9 of p.i. in control mice.Curcumin alone lead to survival of 33% mice whereas group treated with curcumin nanoparticle showed 100% survival. | 113 |
| Albino mice (P. berghei) | Curcuminoid loaded SLN, NLC 100mg/kg | – | IP | Mice treated with drug loaded SLN and NLC showed significantly 2 fold longer survival as compared to control and free curcuminoids. All groups showed 100% mortality at the end of study. | 114 |
| ICR Harlam-Sprague Dawley and C57BL/6 mice (P. berghei ANKA MRA-311/GFP) | 50mg/kg twice a day | 0–5 | IP/Gavage | IP injection of 25mg/kg/d Curcumin on day 1–9 p.i. was beneficial in case of PbA GFP infection in ICR mice; no effect was seen in case of PbA/ICR infection.Gavage treatment of PbA infected C57BL/6 prevented CM and delayed death by 10 days, did not change initial parasitemia and no significant effect was seen in ICR mice. | 115 |
Curcumin has numerous molecular targets in the context of CM. It possesses itself antimalarial activity and acts as a neuroprotective agent:
- i.
NF-κB (nuclear factor κ beta) plays a vital role in malaria as evidenced by PRBCs that have shown to induce NF-κB regulated inflammatory pathway in human cerebral endothelium. The deciphering of anti-malarial activity in curcumin against the blood phase of the life cycle may leads to its role in regulation of NF-κB. Curcumin has profound anti-inflammatory activity and might exert its therapeutic effects by pro-inflammatory cytokine production and expression of cytoadhesion molecules on ECs.100 Curcumin has shown to reduce the production of proinflammatory cytokines such as TNF, IL12 and IL6 in vitro.101 It can be concluded that curcumin can effectively control neuroinflammation and damage due to inflammatory cascade as a result of host immune response and thereby prevent cognitive deficits in CM survivors.
- ii.
Curcumin activates PPAR-γ (peroxisome proliferator activated receptor γ). PPAR-γ is a transcription factor that regulates inflammatory responses. Its activation inhibits translocation of NF-κB and inhibits microglia from producing proinflammatory cytokines.29 It also regulates scavenger receptor CD36, which mediates non-opsonic phagocytosis of PRBCs. Pharmacological upregulation of CD36 in monocytes/macrophages by PPAR-γ agonists has shown to increase CD36 dependent phagocytosis of PRBCs in vitro.102 CD36 is also activated by activation of redox-sensitive transcription factor nuclear related {erythroid-derived 2} factor (Nrf2). These two transcription factors have been found to be activated in macrophages/monocytes treated with curcumin.103 Activation of Nrf2 pathway has been demonstrated as potential target regarding brain inflammation.104
- iii.
It inhibits expression of various cell surface adhesion molecules such as ICAM-1, VCAM-1 and endothelial leukocyte adhesion molecule (ELAM-1) on ECs.94 These data support the development of adjunctive therapies to reverse the pathophysiological consequences of cytoadherence.31 Curcumin also inhibits adhesion of platelets to brain ECs in vitro which accumulate in brain blood vessels in CM patients.105
- iv.
Curcumin causes inhibition of P. falciparum general control nondepressed 5 (PfGCN5). It is a histone acetyl transferase (HAT) that preferentially acetylates K9 and K14 of histone H3. Various post-translational modifications of histone tails lead to regulation of gene expression. Curcumin inhibits HAT and causes parasite chromatin modifications and have antiparasitic effects.99
- v.
Another target of curcumin is PfATP6 (P. falciparum ATP6), the parasite analogue of SERCA (Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase). PfATP6 is the only SERCA-type Ca2+ ATPase in P. falciparum which is responsible for the maintenance of calcium ion concentrations for the generation of calcium-mediated signalling and correct folding and post-translational processing of the proteins.106,107
- vi.
Curcumin inhibits iNOS by suppression of IFN-γ and IL-12 production. iNOS has shown to mediate production of ROS.108
- vii.
Heme oxygenase-1 (HO-1) is the rate limiting enzyme involved in the breakdown of free toxic heme into carbon monooxide, Fe and bilirubin. Curcumin upregulates HO-1 gene and protein expression thereby protecting the ECs from peroxide mediated toxicity as well as toxicity caused due to free heme. Bilirubin further inhibits the ROS production.109,110
- viii.
C-Jun N terminal kinases (JNK) belongs to the family of mitogen activated kinases called MAP Kinases, which are activated in response to inflammatory cytokines and environmental stress conditions. Its activation induces the transcription-dependent apoptotic signalling pathway resulting in neuronal cell death during experimental CM.111 Curcumin suppresses JNK activation and thus protects the neurons
- ix.
Curcumin enhances the activity of detoxifying enzymes like Glutathione-S-transferase.29
All these mechanisms suggest curcumin as an immunomodulator which may alleviate CM.
ConclusionParadoxically the rate of new drug delivery in case of malaria has been very low. The global response to this crisis has been inadequate. There are few new chemical entities in preclinical and clinical phases but these efforts could take considerable time to fructify. So far the attempts have been limited to explore the use of immunomodulator in combination with conventional drugs for CM. Thus, this review highlights the potential of a plant based immunomodulator curcumin as an adjunctive therapy for better management and improvement of survival and overcoming the neurocognitive deficits in CM patients. It may be a useful strategy to complement the direct antiparasitic efficacy of currently used antimalarials and also limit the emergence of resistant parasites.
Conflict of interestThe authors declare no conflicts of interest.
Mr. Kunal Jain is grateful to Council of Scientific and Industrial Research (CSIR), New Delhi, India for financial assistance in the form of Senior Research Fellowship (File No.: 8/484 (0006)/2012-EMR-I). Mr. Sumeet Sood wish to express his gratitude to Department of Science and Technology (DST), New Delhi, India for award of INSPIRE Fellowship (IF10316).
