




Infections, mostly those associated with colonization of wound by different pathogenic microorganisms, are one of the most serious health complications during a medical treatment. Therefore, this study is focused on the isolation, characterization, and identification of microorganisms prevalent in superficial wounds of patients (n=50) presenting with bacterial infection.
MethodsAfter successful cultivation, bacteria were processed and analyzed. Initially the identification of the strains was performed through matrix-assisted laser desorption/ionization time-of-flight mass spectrometry based on comparison of protein profiles (2–30kDa) with database. Subsequently, bacterial strains from infected wounds were identified by both matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and sequencing of 16S rRNA gene 108.
ResultsThe most prevalent species was Staphylococcus aureus (70%), and out of those 11% turned out to be methicillin-resistant (mecA positive). Identified strains were compared with patients’ diagnoses using the method of artificial neuronal network to assess the association between severity of infection and wound microbiome species composition. Artificial neuronal network was subsequently used to predict patients’ prognosis (n=9) with 85% success.
ConclusionsIn all of 50 patients tested bacterial infections were identified. Based on the proposed artificial neuronal network we were able to predict the severity of the infection and length of the treatment.
Skin has an important role in preventing the entry of undesirable substances, organisms, and bacteria into the bloodstream.1,2 Loss of skin integrity leads to different types of wounds which have the humidity, warmth, and a nurturing environment ideal for colonization and proliferation of undesirable bacterial strains, changing the naturally occurring microbiome. Colonized sites are usually polymicrobial, i.e. contain more than one bacterium with pathogenic potential.3 Wound infections are marked by disturbed host–bacteria equilibrium in a traumatized tissue environment favoring the pathogenic bacteria. A wound infection not only has the possibility to elicit a systemic response (sepsis), but is also able to inhibit the multiple processes involved in the orchestrated progression of normal wound healing.4
The concept of microbiome was first suggested in 2001 by Lederberg and McCray and was described as an ecological cohort of commensal, symbiotic, and pathogenic microorganisms sharing a body space.5 Previously, it was estimated that as much as 20 to 60% of human-associated microbiome is hard-to-identify, which has likely resulted in an underestimation of microbiome diversity.6 One of the most frequent microorganisms in infected wound is Staphylococcus aureus.7–10 According to numerous studies,11–13 another common organism in infected wounds is Pseudomonas aeruginosa with up to 10% occurrence, causing nosocomial infections together with S. aureus and other bacteria. In addition, the Enterobacteriaceae family is most often identified in connection with immunocompromised patients or those who have undergone abdominal surgery.1
Bacterial infections, increasingly occurring in medical facilities, can seriously complicate the outcome of treated patients.14,15 This is particularly connected with rising resistance of bacterial strains toward antibiotics or metals,16,17 thus significantly hindering treatment success. Although being highly debated the mechanism of resistance development has not been satisfactorily elucidated.18–21
The elevated occurrence of resistant bacterial strains is strictly linked with increased utilization of invasive surgical techniques, which are often performed in elderly, immunocompromised patients. Simultaneously, with the use of antibiotics, bacterial resistance can evolve in surgical sites, leading to bacteremia and sepsis, and thus significantly prolonging the healing phase of a patient. Although bacterial resistance presents a problem in healthcare facilities, there still exist few possibilities to eliminate the most frequent resistant strains that cause hospital-acquired infection – methicillin-resistant S. aureus (MRSA),22,23e.g. highly potent glycopeptide vancomycin.24 However, for a correct choice of antibiotics one needs to accurately identify the microbiome composition of infected wounds. Knowledge of the bacterial ecology of wounds may thus lead to increase treatment success, coupled with curbing bacterial resistance as a result of inadequate utilization of antibiotics.25–28
Accordingly, this work is focused on identification of the microbiome associated with serious infections in hard-to-heal wounds with the aim to propose a prediction model, comprising both the microbiome composition and patients¿ health conditions.
Materials and methodsChemicals, preparation of deionized water and pH measurementChemicals used in this study were acquired from Sigma-Aldrich (St. Louis, MO, USA) in ACS purity unless noted otherwise. Deionized water was prepared using reverse osmosis equipment Aqual 25 (Aqual s.r.o., Brno, Czech Republic) and further purified using Milli-Q Direct QUV equipped with the UV lamp, with 18MΩ resistance. pH was measured using the pH meter WTW inoLab (Weilheim, Germany).
Preparation of hospital samples and their cultivationCohort of patients with bacterial infectionsFor evaluation, patients with superficial or deep wounds were selected according to infection severity. Detailed information concerning the patients is documented in S1. A total of 50 patients aged 19 through 93 years were enrolled into the clinical study, and 13 patients were 70–79 years old; 23 patients superficial wounds and 27 deep wounds. For all patients, the treatment duration was determined by the traumatologist based severity and extent of infection, associated diseases potentially interfering with treatment outcome and healing of wounds, and other factors such as patient age, concomitant medications, and previous medical history. For confirmation of the functionality of the neural network 9 blank samples from 9 patients identified by MALDI-TOF MS were used. Enrollment of patients into the clinical study was approved by the Ethics Committee of Trauma hospital in Brno.
For evaluation, patients with surficial or deep wounds were selected according to infection severity. Detailed information concerning the patients is documented in S1. A total of 50 patients aged 19 through 93 years were enrolled into the clinical study, and 13 patients were 70–79 years old; 23 patients superficial wounds and 27 deep wounds. For all patients, the treatment duration was determined by the traumatologist based severity and extent of infection, associated diseases potentially interfering with treatment outcome and healing of wounds, and other factors such as patient age, concomitant medications, and previous medical history. For confirmation of the functionality of the neural network 9 blank samples from 9 patients identified by MALDI-TOF MS were used. Enrollment of patients into the clinical study was approved by the Ethics Committee of Trauma hospital in Brno.
Collection of wound swabs from patients with bacterial infectionsThe smears, collected from infected wounds with the agreement of patients, were sampled by rolling motion at the wound using a sterile swab sampler. All patients were divided into two subgroups, on the grounds of infection severity: deep and superficial wound. A detailed description of comorbidities and duration of treatment was obtained. Patients were classified according to the Classification of surgical wounds – SSI (surgical site infections).29–31 Infected wounds were sampled by using disposable tampon swabs maximizing collection of representative microflora. Tampons were subsequently stored in transport medium (inorganic salts, sodium thioglycolate, 1% agar, activated charcoal). The important part of our workflow process comprised sampling in duplicates with further transport in both aerobic and anaerobic conditions to preserve bacterial viability.
Cultivation of clinical specimensFour types of selective nutrient media (blood agar enriched by 10% NaCl, Endo agar, blood agar without any other component, and blood agar with amikacin) we employed for further microbiological selection. Petri dishes, containing the above mentioned media were subsequently incubated according to conventional protocols, as described elsewhere,32–35 to maintain suitable conditions for growth of all types of bacteria. These Petri dishes were incubated for 24–48h at 37°C supplemented by TGY medium (1gL−1 glucose, 5gL−1 tryptone, 2.5gL−1 yeast extract). Subsequently, individual colonies were collected from each Petri dish and stored in 1μL of enriched media. These samples were processed as described in the DNA sequencing section and utilized for both – MALDI-TOF MS identification and PCR with subsequent sequencing.
MALDI-TOF MS identification of bacteriaThe following extraction protocol was based on MALDI Biotyper™ 3.0 User Manual Revision 2, also used in a previous report.36 500μL of bacterial culture, cultivated overnight, was centrifuged at 14,000×g for 2min. The supernatant was discarded and the pellet was re-suspended in 300μL of deionized water besides adding 900μL of ethanol. After centrifugation at 14,000×g for 2min, the supernatant was discarded and the obtained pellet was air-dried. The pellet was then dissolved in 25μL of 70% formic acid (v/v) and 25μL of acetonitrile and mixed. The samples were centrifuged at 14,000×g for 2min and 1μL of the clear supernatant was spotted in duplicate onto the MALDI target and air-dried at room temperature. Then, each spot was overlaid with 1μL of α-cyano-4-hydroxycinnamic acid (HCCA) matrix solution (20mgmL−1) in organic solvent (50% acetonitrile and 2.5% trifluoroacetic acid, both v/v) and air-dried completely prior to MALDI-TOF MS measurement on UltrafleXtreme MS (Bruker Daltonik GmbH, Bremen, Germany). As matrix solution 2,5-dihydroxybenzoic acid (DHB) was also used in the same concentration and solvent as HCCA. Spectral data were taken in the m/z range of 2000–30,000Da, resulted from the accumulation of 240 laser shots targeted to different regions of the same sample spot. These data were analyzed with the Flex Analysis software (Version 3.4). Final preparation of dendrograms was carried out in the MALDI BioTyper™ 3.1 (Bruker Daltonik GmbH, Bremen, Germany).
DNA sequencingBacterial cells were centrifuged at 4450×g and 20°C for 10min. The pellet was resuspended in 400μL of lysis buffer (6M guanidium hydrochloride, 0.1M sodium acetate) and cells were lysed for 1hour at 20°C and 600rpm on Multi RS-60 (Biosan, Riga, Latvia). Genomic DNA was isolated from lysed bacterial cultures via MagNA Pure Compact (Roche, Mannheim, Germany), using Nucleic Acid Isolation Kit I, protocol DNA_Bacteria.
16S rRNA gene was amplified using Taq PCR Mix (New England Biolabs, Ipswich, MA, USA) and MasterCycler realplex4 epgradient S (Eppendorf, Hamburg, Germany). 100μL of reaction mixture consisted of: 1× Standard Taq Reaction Buffer, 1.6U of Taq DNA polymerase, 200μM Deoxynucleotide Solution Mix, 0.5μM primers and 5μL of isolated genomic DNA. The forward primer E9F 5′-GAGTTTGATCCTGGCTCAG-3′ and reverse primer U1510R 5′-GGTTACCTTGTTACGACTT-3′ were synthesized by Sigma–Aldrich (St. Louis, MO, USA). The reaction profile was as follows: initial denaturation at 94°C for 4min; 30 cycles of denaturation at 94°C for 30s, annealing at 52°C for 30s and elongation at 72°C for 105s; with terminal elongation at 72°C for 10min. Amplified fragments were purified using MinElute PCR Purification Kit (Qiagen, Hilden, Germany).
For sequencing reaction the DTCS Quick Start Kit (Beckman Coulter, Pasadena, CA, USA) was used. To 20μL sequencing reaction mixture, 98ng of purified fragment, 0.75μL of 10μM forward primer, 4μL of DTCS Quick Start Master Mix and 1μL of Sequencing Buffer were added. The conditions of 30 cycle-reactions were as follows: 96°C for 20s; 50°C for 20s and 60°C for 4min. The purification of sequencing product was carried out using CleanSEQ kit (Beckman Coulter). Purified samples in Sample Loading Solution were transferred to the plate and DNA sequencing was performed using Genetic Analysis System CEQ 8000 (Beckman Coulter). After denaturation at 90°C for 2min, a fluorescence-marked DNA fragments were separated in 33cm long capillary with 75μm i.d. (Beckman Coulter) filled with linear polyacrylamide denaturing gel. The separation was run at capillary temperature of 50°C and voltage of 4.0kV for 85min. Sequences were identified by comparison with NCBI database.
Amplification of S. aureus genes mecA and fnbAIsolation of genomic DNA was performed using the same method as described in section DNA sequencing. The mecA and fnbA genes were amplified using polymerase chain reaction (PCR) as previously reported.37 The primers were synthesized by Sigma-Aldrich and the sequences of forward and reverse primers for mecA gene were 5′-CCCAATTTGTCTGCCAGTTT-3′, and 5′-TGGCAATATTAACGCACCTC-3′ and for fnbA gene were 5′-GATACAAACCCAGGTGGTGG-3′, and 5′-TGTGCTTGACCATGCTCTTC-3′. The volume of PCR reaction mixture was 100μL containing 1× Taq reaction buffer, 0.2mM dNTP, 1.6U of Taq DNA polymerase (New England Biolabs) and 0.5mM of each primer. The reaction profile was as follows: initial denaturation at 94°C for 4min, 30 cycles of denaturation at 94°C for 30s, annealing at 53°C for 30s and extension at 72°C for 1min with a final extension of 7min. The amplification generated a 223bp for mecA and 191bp for fnbA.
Agarose gel (2% (v/v), high melt, Mercury, San Diego, CA, USA) was prepared with 1× TAE buffer (40mM Tris, 20mM acetic acid and 1mM ethylenediaminetetraacetic acid) and ethidium bromide (5μL per 100mL of the gel) as described elsewhere.38 100bp DNA ladder (New England Biolabs) within the size range from 100 to 1517bp was used to monitor the size of the analyzed fragment. The electrophoresis (Bio-Rad, Hercules, CA, USA) was run at 60V and 6°C for 160min. The bands were visualized by UV transilluminator at 312nm (Vilber-Lourmant, Marne-la-Vallée, France).
Statistical processing of obtained resultsAutomated neuronal network was used as a predictive model. Classification analysis automated neuronal network was used for the estimation of categorical data. The dataset was randomly divided as follows: 80% for learning, 10% for testing, and 10% for validation. Following network types were tested using automated network search: multilayer perceptron network (MLP), and radial basis function (RBF). Number of hidden units to search was determined as follows: 8–24 and 8–11 for MLP and RBF, respectively. Total 1000 networks were trained, and activation functions were searched for identity, logistic, tanh, exponential. Weight decay of 0.0001–0.001 was used for hidden layer and output layer. Weight of input variables for learning was used based on MALDI-TOF classification score. Unless noted otherwise, p-value less than 0.05 was considered significant. Software Statistica 12 (StatSoft, CA, USA) was used for analysis.
Results and discussionWe decided to employ a variety of cultivation approaches (in presence of O2, CO2 or in microaerophilic conditions) to reveal the presence of real microbiota associated with superficial infections.
MALDI-TOF MS was explored as an accurate and rapid identification tool, using the protein mass patterns, which are compared with patterns from a commercial Bruker Daltonics database (BDAL) of MALDI Biotyper™ software.39 Due to a powerful software support, the method can be used for identification within few minutes, which is one of the advantages.36 Moreover, sequencing of amplified 16S rRNA gene40 was employed for identification independent of protein patterns. Finally, an artificial neural network (ANN) was developed as a predictive model for evaluation of infection severity and using developed ANN we attempted to find the relationship between disease severity and the microorganisms identified in clinical specimens.
Identification of bacterial strains by MALDI-TOF MS and Sanger sequencingFor the identification of bacterial entities we employed complementary methods for independent evaluation of different biomolecules – proteins and DNA.36,41,42 Sanger sequencing was utilized as a confirmation method, based on sequencing of 16S rRNA gene. This gene contains hypervariable regions, providing species-specific sequences, hence it can provide enough information for a confident discrimination, and thus became popular in medical microbiology to classify bacteria.43,44
When compared to sequencing, MALDI-TOF MS offers much shorter analysis time. By using this technique, wound microbiome could be discriminated within one hour of incubation, and thus this will likely become the method of choice for future microbiome identification. Nevertheless, the classification is based on a still developing database34; hence MALDI-TOF MS identification of non-databased bacteria has still to be connected with other confirmation methods. From this reason we firstly employed MALDI-TOF MS with a condition: If score <2.00=16S rRNA sequencing.
As shown in S2, 108 bacterial strains were identified37 of them had to be confirmed by sequencing and confirmed strains were immediately databased to increase future classification success. Strains of S. aureus were the most often identified (n=35). Thus, methicillin-resistant S. aureus (MRSA) is highly associated with severe infections in post-surgical wounds45; we further analyzed the mecA gene, encoding a modified penicillin binding protein (PBP) known as PBP2a, with decreased affinity toward β-lactams.46 The mecA positivity was determined in four isolates. Since 67% of patients had deep wound infections and were treated for more than 8 weeks after admission to infectious Department of Trauma Hospital of Brno, presence of mecA was shown to be a crucial microbiological factor, affecting patients prognosis. Further, we determined the presence of fnbA gene, responsible for adhesins production. Adhesion to human extracellular matrix components and serum proteins is an important facet in the interaction between bacteria and its host cells.47 Lim and coworkers identified the presence of fnbA in 96% of all isolated MRSA strains.35 In our case, fnbA presence was confirmed in all MRSA isolates and in 89% of methicillin-sensitive S. aureus isolates. Similarly to mecA, fnbA was found to be associated with infection severity. In patients with negative fnbA and mecA the treatment duration was less than four weeks in 75% of cases, despite the fact that patients had deep wound infections. This finding suggests that the severity of staphylococcal infections does not depend solely on antimicrobial resistance, but also on adhesins expression, which enhance the interaction with the target host cells.
Distribution of identified strains within various cohorts of patientsAccording to duration of treatment, the patients were divided into specific subgroups, where each sector represents one bacterial strain.
The subdivision of patients was based on surgical wounds classification SSI. As shown, patients were divided into two groups - deep and superficial wounds and the associated bacterial strains are depicted in Fig. 1A and B.
 Infection severity – deep wounds and (B) infection severity – superficial wounds.")
As it is obvious from Fig. 1A, in the more serious infections (deep) S. aureus was the main bacterium of microbiome composition (28% of identified strains), followed by Enterococcus faecalis (15%), and Escherichia coli (11%). On the other hand, E. coli was not so often identified in surficial wounds (5% – Fig. 1B). Taken together, the microbiome composition in both groups exhibits substantial differences, and thus it can be hypothesized that presence of minority representatives as Hafnia alvei, Proteus vulgaris, Staphylococcus lugdunensis, or Enterobacter cloacae in the wound can significantly influence the infection severity. It can be also stated that increasing duration of treatment leads to increased number of identified Enterobacteriaceae and opportunistic pathogens (Pseudomonas, Enterococcus).
Phylogenetic analysis of protein alterationsAs was shown by Rettinger and colleagues,48 MALDI-TOF mass spectra-based phylogenetic analysis is considered equivalent to 16S rRNA gene sequencing. Therefore, we employed MALDI Biotyper™ for preparation of dendrograms for our groups, divided by treatment duration (Fig. 2). Dendrograms showed similarity of same bacterial strains (low distance level), but in some cases larger differences were found – usually among bacterial strains from different patients. These differences were caused probably by modifications of bacterial proteins. Karger et al. found methylation as a cause of higher distance level in dendrogram between Burkholderia pseudomallei and other types of B. pseudomallei.49 Thus it can be concluded that not only changes in microbiome representatives affect treatment duration and success, but also small changes in protein post-translation modifications can be highly important for patients’ recovery.
Artificial neural network Treatment duration less than four weeks. (B) Treatment duration 4–7 weeks. (C) Treatment duration eight and more weeks. (D) Exitus.")
Two neuronal networks were created: (1) for the prediction of time-to-heal, and (2) for the prediction of infection severity. The following input parameters were used for the construction of networks: from 2000 networks five were retained and one was used for further final custom neuronal network. The settings of the network created using automated algorithm and used for the custom final learning were Multilayer perceptron 89-13-3 (input-hidden-output neurons), Broyden–Fletcher–Goldfarb–Shanno (BFGS) training algorithm, sum of squares error function, identity function for hidden layer, and then for output layer. The design of the network is displayed in Fig. 3A. With stopping conditions enabled (Fig. 3B), a final network was created in the 17th training cycle with performances of 91.4%, 85.7%, and 71.4% for training, testing, and validation (accuracy in prediction up to 85% – Fig. 3C), respectively.
 Design of classification network for the prediction of time-to-heal. The number of neurons/inputs is indicated by n. *Note the number of input and hidden neurons is not displayed exactly. (B) Training process of the classification network with stopping conditions activated. (C) Accuracy of the final network for classification of time-to-heal. (D) Design of classification network for the prediction of infection severity. (E) Training process for creation of this network with stopping criteria activated. (F) Accuracy of the network for the prediction of infection severity.")
Design and performance of the neuronal networks. (A) Design of classification network for the prediction of time-to-heal. The number of neurons/inputs is indicated by n. *Note the number of input and hidden neurons is not displayed exactly. (B) Training process of the classification network with stopping conditions activated. (C) Accuracy of the final network for classification of time-to-heal. (D) Design of classification network for the prediction of infection severity. (E) Training process for creation of this network with stopping criteria activated. (F) Accuracy of the network for the prediction of infection severity.
Consequently, a second neuronal network for the prediction of infection severity was created using an automated algorithm. The best-performing network was trained under following settings: multilayer perceptron 89-19-2 (input-hidden-output neurons) (Fig. 3D) BFGS training algorithm, cross entropy error function, and exponential and softmax activation function for hidden and output layer. The training process is depicted in Fig. 3E (accuracy in prediction up to 85% – Fig. 3F).
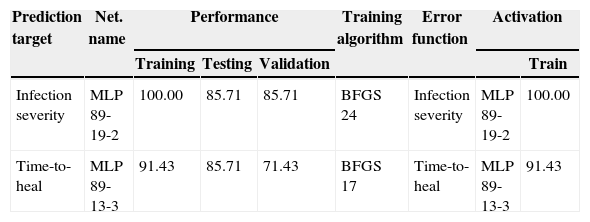
The performances of the network were 100.0%, 85.7%, and 85.7% for training, testing, and validation (Table 1), respectively. The accuracy for individual cases is displayed in Table 2.
Characterization of neuronal network performance for the prediction of patient outcome. Performance displayed in % for training, testing, and validation samples. The number of training cycle for custom network training is displayed in training algorithm column. BFGS, Broyden–Fletcher–Goldfarb–Shanno training algorithm; SOS, sum of squares.
| Prediction target | Net. name | Performance | Training algorithm | Error function | Activation | |||
|---|---|---|---|---|---|---|---|---|
| Training | Testing | Validation | Train | |||||
| Infection severity | MLP 89-19-2 | 100.00 | 85.71 | 85.71 | BFGS 24 | Infection severity | MLP 89-19-2 | 100.00 |
| Time-to-heal | MLP 89-13-3 | 91.43 | 85.71 | 71.43 | BFGS 17 | Time-to-heal | MLP 89-13-3 | 91.43 |
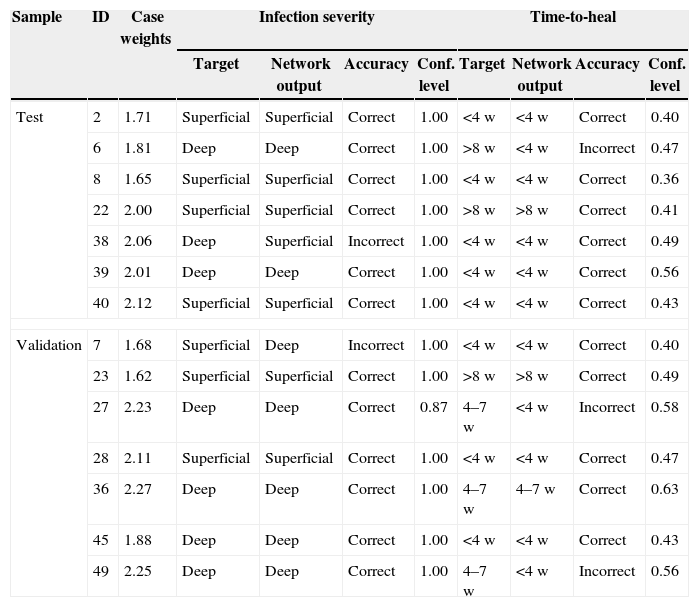
Performance of the network: verification of the test and validation cohort. Analysis for both networks for prediction of infection severity and time-to-heal. Test cohort was employed for stopping conditions. Validation sample was used to test final network. “target” indicates input data, network output reflects calculated result from the neuronal network. id, identification of patient; w, week.
| Sample | ID | Case weights | Infection severity | Time-to-heal | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Target | Network output | Accuracy | Conf. level | Target | Network output | Accuracy | Conf. level | |||
| Test | 2 | 1.71 | Superficial | Superficial | Correct | 1.00 | <4w | <4w | Correct | 0.40 |
| 6 | 1.81 | Deep | Deep | Correct | 1.00 | >8w | <4w | Incorrect | 0.47 | |
| 8 | 1.65 | Superficial | Superficial | Correct | 1.00 | <4w | <4w | Correct | 0.36 | |
| 22 | 2.00 | Superficial | Superficial | Correct | 1.00 | >8w | >8w | Correct | 0.41 | |
| 38 | 2.06 | Deep | Superficial | Incorrect | 1.00 | <4w | <4w | Correct | 0.49 | |
| 39 | 2.01 | Deep | Deep | Correct | 1.00 | <4w | <4w | Correct | 0.56 | |
| 40 | 2.12 | Superficial | Superficial | Correct | 1.00 | <4w | <4w | Correct | 0.43 | |
| Validation | 7 | 1.68 | Superficial | Deep | Incorrect | 1.00 | <4w | <4w | Correct | 0.40 |
| 23 | 1.62 | Superficial | Superficial | Correct | 1.00 | >8w | >8w | Correct | 0.49 | |
| 27 | 2.23 | Deep | Deep | Correct | 0.87 | 4–7w | <4w | Incorrect | 0.58 | |
| 28 | 2.11 | Superficial | Superficial | Correct | 1.00 | <4w | <4w | Correct | 0.47 | |
| 36 | 2.27 | Deep | Deep | Correct | 1.00 | 4–7w | 4–7w | Correct | 0.63 | |
| 45 | 1.88 | Deep | Deep | Correct | 1.00 | <4w | <4w | Correct | 0.43 | |
| 49 | 2.25 | Deep | Deep | Correct | 1.00 | 4–7w | <4w | Incorrect | 0.56 | |
Sensitivity analysis of input variables for both networks was carried out. For the prediction of infection severity, the mean sensitivity level was 4.66, ranging from 0.63 to 20.1, and a total sensitivity=179.9 (Fig. 4) The highest level of sensitivity (thus highest impact on prediction of a network) was observed for hypertension (20.15), ulcerative colitis (17.06), obesity (10.13), E. coli (8.87), E. faecalis (8.50), and other factors. The factors with sensitivity <1 were P. vulgaris, Neisseria mucosa, E. cloacae, S. aureus 2 mecA genes, Staphylococcus haemolyticus, paresis/plegia, Alzheimer's disease, age.
 Sensitivity of individual factors for the prediction of time-to-heal. (B) Sensitivity of individual factors for the prediction of infection severity.")
Sensitivity analysis of all factors for prediction of time-to-heal and infection severity. Sensitivity of individual factors depicted as a percentage of total sensitivity. (A) Sensitivity of individual factors for the prediction of time-to-heal. (B) Sensitivity of individual factors for the prediction of infection severity.
For the prediction of time-to-heal, the sensitivity was distinctly more homogeneous for the input factors with mean sensitivity=1.35, (0.99–2.35), total sensitivity=52.49. The factors with higher sensitivity included ulcerative colitis (2.35), Parkinson's disease (2.27), osteomyelitis (2.15), Pseudomonas mendocina (2.11), S. haemolyticus (1.84) and others (Fig. 4). The factors with sensitivity <1 include Streptococcus agalactiae, Corynebacterium striatum, and Stenotrophomonas maltopilia.
Conflicts of interestThe authors declare no conflicts of interest.
The study was financially supported by CEITECCZ.1.05/1.1.00/02.0068. The authors wish also to express their special thanks to Radek Chmela for perfect technical assistance.
The following are the supplementary data to this article:
