



Hepatitis B virus (HBV) is distributed worldwide, with geographical variations regarding prevalence of the different genotypes. The aim of this study was to determine the HBV genotypes and subgenotypes circulating in Southeast Brazil and compare the genetic sequences found with HBV sequences previously described in the world. Sequences from 166 chronic HBV carriers were analyzed using the fragment constituted by 1306 base pairs comprising surface and polymerase regions of the HBV genome. The sequences obtained were submitted to phylogenetic analysis. HBV subgenotypes A1, A2, D1-D4, F2a, and F4 were found. HBV genotype D was the most frequent, found in 99 patients (58.4%). Within this group, subgenotype D3 was the most prevalent, in 73 patients (42.9%). HBV genotype A was identified in 58 (36%) patients, subgenotype A1, in 48 (29.8%) subjects. Genotype F was identified in 9 (5.4%). According to the phylogenetic analysis, the sequences found were grouped with sequences from Europe, Asia and Middle East (subgenotypes D1, D2, D3) and sequences from Latin America and Africa (subgenotype A1). HBV D3 grouped in different clusters inside D3 clade, several of them with sequences isolated in Italy. We also identified eight families whose relatives were infected with the same HBV subgenotype, most with high similarity between sequences. In conclusion, the distribution of the HBV sequences obtained interweaved with sequences from other continents, corresponding to regions from where many immigrants came to this region, in accordance to the hypothesis that the HBV detected over there were brought during the colonization times.
The genotype and subgenotype characterization of hepatitis B virus (HBV) contributes to understanding the natural history of infection, since genetic heterogeneity of the virus seems to imply in biological properties which may influence the clinical outcomes and the response to antiviral treatment. Moreover, phylogenetic analysis of HBV genome sequences helps understanding the epidemiology of this infection and the virus spread through a given geographical region. HBV is distributed worldwide with differences regarding its endemicity and frequency of the different genotypes and subgenotypes.1 Although they probably share a common ancestor, over time there have been different evolutionary histories for the spread of HBV in distinct geographic areas. It is believed that population migratory processes have modified the distribution of HBV genotypes and subgenotypes throughout the world.1 Brazil is a country of continental dimensions, composed by a heterogeneous population in relation to ancestral ethnic contributions. Studies in population subgroups have described the prevalence of HBV genotypes circulating in some regions of the country, particularly in the capital cities and in the Amazon basin. The most frequent HBV genotype encountered was A, followed by genotypes D and F.2–7 Ribeirão Preto region is located in São Paulo, the most populous and largest gross domestic product per capita state in Brazil (Fig. 1). The population composition of this region was strongly influenced by the coffee agriculture in the nineteenth century, which contributed to the massive Italian migration, unlike other regions of the country, especially toward the north-northeast, where the Portuguese colonization, the presence of natives and Africans brought into slavery gave rise to the miscegenation that makes up the current population. Ribeirão Preto region were always subject to intense migratory process, as it maintained a rapid growth rate in the twentieth century and still retained the status of an attracting region in socio-economic terms for migrants from other country regions as well as from abroad.

The aim of this study was to determine the HBV genotypes and subgenotypes circulating in the Ribeirão Preto region and to compare the genetic sequences found with HBV sequences previously described in different parts of the world by phylogenetic analysis.5–9
Material and methodsThis was a cross-sectional study.
Study subjectsBetween July 2007 and July 2009, HBV chronically infected patients were recruited from the Hepatitis B Outpatient Clinic of the Clinical Hospital of the Faculty of Medicine of Ribeirão Preto, University of São Paulo (HC FMRP USP). Inclusion criteria for this study were age above 18 years, presence of HBsAg in serum for more than six months at the time of the study initiation. The exclusion criteria were as follows: co-infection with hepatitis C virus (HCV) or the human immunodeficiency virus (HIV), and causes of liver disease other than HBV. On the day of enrollment, patients underwent an interview, physical examination, review of medical records, and had blood drawn for laboratorial analysis. Information about first-degree relatives infected by HBV also included in this study were registered.
The study was approved by the Research Ethics Committee of the Hospital (authorization No. 1809/2007) and all patients gave written informed consent to participate. The diagnosis of cirrhosis was based on clinical features (ascites, spiders, encephalopathy, etc.), biochemical and coagulation tests, liver and portal system doppler ultrasonography, upper gastrointestinal endoscopic parameters, or in the presence of characteristic histopathological features observed in a liver biopsy fragment. The diagnosis of hepatocellular carcinoma (HCC) was defined according to the Barcelona-Clinic Liver Cancer Group.8
Serological analysisEnzyme Linked Immuno Sorbent Assay (ELISA) was used for the detection of HBV antigens and antibodies: HBsAg, HBeAg, anti-HBc IgG, anti-HBs, anti-HBe (Axsym, ABBOTT) and anti-HDV (ETI-AB-DELTAK-2, DiaSorin, Italy) antibodies.
HBV DNA quantification and genotype/subgenotype characterizationThe viral load was determined as previously described9 and samples with detectable HBV DNA were submitted to nested polymerase chain reaction using primers that amplify a 1306 pb fragment of the surface and polymerase (S/Pol) genes.10 Amplified PCR fragments were purified using the Charge Switch PCR Clean Up Kit (Invitrogen), quantified and then sequenced by the Sanger methodology using dideoxynucleotides (ddNTPs) and the ABI PrismR BigDyeTM Terminator Kit and three pairs of primers that generated six sequences (three sense and three antisense) with approximately 500 base pairs overlapping.10 Sequencing was performed in ABI 3500 automated DNA sequencer (Applied Biosystems, Thermo Scientific Foster City, CA, USA). Sequences obtained were submitted to quality evaluation using the program Phred-Phrad and the consensus sequences were assembled by alignment of the six sequences of each sample using CAP3 software available in Electropherogram quality analysis site (http://asparagin.cenargen.embrapa.br/phph/).
HBV genotypes and subgenotypes were identified by phylogenetic analysis of the sequences characterized in this study together with reference sequences available in GenBank. To perform this analysis the sequences were initially aligned using the Clustal_W program integrated in BioEdit software,11 which was also used to alignment editing, then a Bayesian analysis was conducted using BEAST v1.8.3 software.12 The phylogeny was estimated using GTR+G+I as nucleotide substitution model (selected using jModelTest 2.1.10)13 under relaxed (uncorrelated lognormal) molecular clock. Markov Chain Monte Carlo (MCMC) was run for 10 million generations with sampling every 1000 steps. Trees were summarized in a maximum clade credibility tree using TreeAnotator v 1.8.3, with 10% burn-in and then visualized and edited using FigTree v1.4.2 (Available online: http://tree.bio.ed.ac.uk/). Additionally, HBV transmission among individuals belonging to the same family nucleus was evaluated by analysis of the clustering pattern of the HBV strains isolated from each individual.
Statistical analysisResults were analyzed using the statistic program GraphPadPrism (Software Inc., San Diego, CA, version 5.01). Descriptive statistics are presented as means, medians, and standard deviations (SD), and Qui Square Test and Fischer's Exact Test when indicated were used to compare categorical variables. The significance level for all statistical tests was p≤0.05.
ResultsThe study was conducted in a tertiary reference health service, located in the municipality of Ribeirão Preto, São Paulo state, Southeastern region of Brazil (Fig. 1). There were 193 subjects initially included in the study. It was possible to amplify and sequence the S/POL HBV region from 166 patients, as in the remaining 27 HBV DNA was not detected. A total of 104 patients were male (64.6%), with ages ranging from 19 to 77 years, mean 44.1 (SD=12.2) and median 43 years. All the patients included were anti-HDV negative. Of the total, 24 patients (14.4%) had hepatic cirrhosis and 14 (8.4%) HCC.
Only three HBV genotypes were found in this study sample, genotype D (59.6%; 99/166) and A (35%; 58/166) being the most prevalent ones with few cases infected with genotype F (5.4%; 9/166). Considering subgenotypes, a high genetic diversity was observed: subgenotypes A1, A2, D1–D4, F2a and F4 were found (Figs. 2 and 3). The subgenotype D3 was the most prevalent (73.7%; 73/99) among the patients infected by genotype D, followed by D2 (14%; 14/99), D1 (8%; 8/99) and D4 (4%; 4/99); HBV/A1 was the main subgenotype of genotype A (82.7%; 48/58) and among those patients infected with genotype F, subgenotype F2a was the most frequent (77.7%; 7/9).
 Phylogenetic trees of the sequences identified in this study, classified as genotype D: Subgenotype D3. (B) Subgenotypes D1, D2 and D4. Only sequences belonging to these subgenotypes where shown individually, all the other ones were collapsed. The analysis was conducted using a Bayesian analysis using BEAST v1.8.3 software with 296 sequences (GenBank 197 and 99 of this study) of 1270 nucleotides of S/polymerase in the HBV genome. Probability posterior values are shown in the branches. The sequences characterized in this study are marked with the symbol # and the sequences taken from GenBank are identified by subgenotype, followed by the access number and geographical origin. Sequences from members of the same family were shown with a circle and each family is identified by F and a number (F1–6 and F8).")
 Phylogenetic trees of the sequences identified in this study, classified as genotype D: Subgenotype D3. (B) Subgenotypes D1, D2 and D4. Only sequences belonging to these subgenotypes where shown individually, all the other ones were collapsed. The analysis was conducted using a Bayesian analysis using BEAST v1.8.3 software with 296 sequences (GenBank 197 and 99 of this study) of 1270 nucleotides of S/polymerase in the HBV genome. Probability posterior values are shown in the branches. The sequences characterized in this study are marked with the symbol # and the sequences taken from GenBank are identified by subgenotype, followed by the access number and geographical origin. Sequences from members of the same family were shown with a circle and each family is identified by F and a number (F1–6 and F8).")
(A) Phylogenetic trees of the sequences identified in this study, classified as genotype D: Subgenotype D3. (B) Subgenotypes D1, D2 and D4. Only sequences belonging to these subgenotypes where shown individually, all the other ones were collapsed. The analysis was conducted using a Bayesian analysis using BEAST v1.8.3 software with 296 sequences (GenBank 197 and 99 of this study) of 1270 nucleotides of S/polymerase in the HBV genome. Probability posterior values are shown in the branches. The sequences characterized in this study are marked with the symbol # and the sequences taken from GenBank are identified by subgenotype, followed by the access number and geographical origin. Sequences from members of the same family were shown with a circle and each family is identified by F and a number (F1–6 and F8).
 Phylogenetic tree of the sequences identified in this study, classified as genotype A: Subgenotype A1. (B) Subgenotypes A2, F2A and F4. Only sequences belonging to these subgenotypes where shown individually, all the other ones were collapsed. The analysis was conducted using a Bayesian analysis using BEAST v1.8.3 software with 328 sequences (GenBank 261 and 67 of this study) of 1270 nucleotides of the S/polymerase in the HBV genome. Probability posterior values are shown in the branches. The sequences characterized in this study are marked with the symbol # and the sequences taken from GenBank are identified by subgenotype, followed by the access number and geographical origin. Sequences from members of the same family were shown with a circle and the family 7 is identified as F7.")
(A) Phylogenetic tree of the sequences identified in this study, classified as genotype A: Subgenotype A1. (B) Subgenotypes A2, F2A and F4. Only sequences belonging to these subgenotypes where shown individually, all the other ones were collapsed. The analysis was conducted using a Bayesian analysis using BEAST v1.8.3 software with 328 sequences (GenBank 261 and 67 of this study) of 1270 nucleotides of the S/polymerase in the HBV genome. Probability posterior values are shown in the branches. The sequences characterized in this study are marked with the symbol # and the sequences taken from GenBank are identified by subgenotype, followed by the access number and geographical origin. Sequences from members of the same family were shown with a circle and the family 7 is identified as F7.
The phylogenetic relationships among HBV sequences characterized here with sequences isolated in other countries available in GenBank were assessed. This analysis showed that HBV/D3 circulating in Ribeirão Preto, Brazil, grouped in different clusters inside D3 clade and several of them have sequences isolated in Italy (Fig. 2A).
HBV/D1 and D2 showed the same grouping pattern and are closely related to sequences from Europe, Middle East, and Asia (Fig. 2B). Concerning subgenotype D4, we observed that the sequences grouped in a different clade of D4 sequences previously characterized in other regions of Brazil, and one of this sequences cluster near a clade of sequences isolated mainly in other countries of Latin America (Haiti, Cuba, and Martinique). Interestingly, there was a highly supported branch including other subgenotype D4 sequences from Brazil obtained from Maranhão (Northeast region) and Rondônia (North region) that did not include any sequence from Ribeirão Preto (Fig. 2B).
HBV/A1 sequences found in Ribeirão Preto grouped in different clusters inside Asian/America A1 clade and most of them were close related with A1 sequences isolated in other Brazilian cities or in other countries of Latin America (Fig. 3A). HBV/A2 sequences did not cluster with sequences from any particular origin. The few F2a sequences grouped together with other samples obtained in Rondônia, whereas HBV/F4 grouped with sequences from Argentina and Bolivia (Fig. 3B).
HBV subgenotypes distribution was analyzed according to the origin of the patients included in this study. A total of 157 patients (94.6%) lived in the Ribeirão Preto municipality or in nearby cities, 100km distant at most. Of these patients, 45.8% (n=72) were infected with HBV subgenotype D3, 26.1% (n=41) with HBV subgenotype A1 and the remaining with the following subgenotypes: D2 (8.9%; n=14), A2 (5.7%; n=9), D1 (3.8%; n=8), F2a (4.0%; n=7), D4 (2.0%; n=4), and F4 (1.3%; n=2). Among the remaining nine patients (from cities up to 300km from Ribeirão Preto, within São Paulo state or from other states) most (77.7%; 7/9) were infected with HBV/A1, one with HBV/A2, and other with HBV/D3.
HBV subgenotypes and clinical outcome were evaluated. Among the 24 patients with hepatic cirrhosis, 13 (54.2%) presented HBV genotype A infection (50% HBV subgenotype A1 and 4.2% HBV subgenotype A2), 10 (41.7%) HBV genotype D (4.2% subgenotype D2, 33.3% D3, 4.2% D4), and one patient (4.2%) HBV/F2a. Among the 14 patients with HCC, seven (50%) presented HBV genotype A infection (all HBV subgenotype A1); six (42.9%) were infected by HBV genotype D (7.1% HBV/D2, 28.6% HBV/D3, 7.1% HBV/D4), and one (7.1%) presented HBV/F2a infection. There was no association between the different HBV genotypes or subgenotypes and hepatic cirrhosis (p=0.10 and p=0.35 respectively), neither the presence of HCC (p=0.43 and p=0.42 respectively).
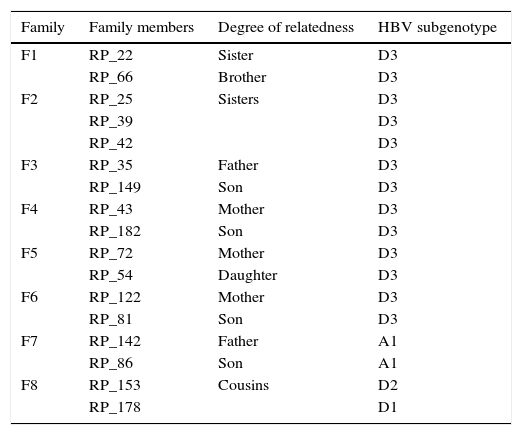
Patients from eight different families were identified in this study and HBV sequences isolated from them were used to evaluate the occurrence of intrafamilial transmission. Table 1 shows the family relationship between the patients and HBV subgenotype identified in each case. Very closely related HBV isolates were observed mainly when relatives were mother and children (families F4, F5, and F6). In these cases high similarity between sequences represented by grouping pattern in the same branch of the phylogenetic tree with high probability posterior value were observed (Fig. 2). All members of these families were infected with HBV/D3.
Patients grouped with family members and HBV genotypes and subgenotypes.
| Family | Family members | Degree of relatedness | HBV subgenotype |
|---|---|---|---|
| F1 | RP_22 | Sister | D3 |
| RP_66 | Brother | D3 | |
| F2 | RP_25 | Sisters | D3 |
| RP_39 | D3 | ||
| RP_42 | D3 | ||
| F3 | RP_35 | Father | D3 |
| RP_149 | Son | D3 | |
| F4 | RP_43 | Mother | D3 |
| RP_182 | Son | D3 | |
| F5 | RP_72 | Mother | D3 |
| RP_54 | Daughter | D3 | |
| F6 | RP_122 | Mother | D3 |
| RP_81 | Son | D3 | |
| F7 | RP_142 | Father | A1 |
| RP_86 | Son | A1 | |
| F8 | RP_153 | Cousins | D2 |
| RP_178 | D1 |
On the other hand, in the remaining families it was not possible to confirm the occurrence of intrafamilial HBV transmission because HBV sequences isolated from its members were distributed in three different branches (families F2, F7, and F8), or on the same branch together with sequences isolated from patients that were not members of the family (families F1, F2, and F3) (Fig. 3).
DiscussionUsing phylogenetic analysis, several studies conducted in different parts of the world have shown that HBV migrates with its hosts, so the distribution of the different genotypes and subgenotypes throughout the continents and among the several regions within each continent is the result of these migrations.
The consequence of migrations from Europe and Africa that occurred during the colonial period in the actual picture of HBV genotype/subgenotypes distribution in Brazil has been proposed and discussed by several studies. The introduction of HBV/A1 and HDV/D4 has been attributed to African slavery brought to Brazil2,14,15 whereas the introduction of HBV/D2 and HBV/D3 has been related to European immigration.16,17 On the other hand the presence of HBV/F2a has been related to presence of indigenous natives, which currently most of them live in the north region of the country.18
HBV genotypes A (A1 and A2), D (D1, D2, D3 and D4) and F (F2a and F4) are the mainly HBV genotypes found in different Brazilian regions studied until now. HBV/A (mainly A1) prevails in North, Northeast, and Southeast regions and HBV/D (mainly D2 and D3) in the South region.10,14–19 Although HBV genotype A has been the most common genotype found in different cities of the Southeast region,18,20–24 in Ribeirão Preto we found a higher (59.6%; 99/166) prevalence of genotype D, represented mainly by subgenotype D3 (73.7%; 73/99). Interestingly, when the association between HBV subgenotypes and origin of the patients was evaluated most (45.8%) of the cases who lived in Ribeirão Preto or municipality/nearby cities were infected with HBV/D3. On the other hand, those living in cities within São Paulo state up to 300km from Ribeirão Preto or from other Brazilian states were more frequently (77.7%) infected with HBV/A1, suggesting that HBV/D3 is a subgenotype characteristic of this region.
The pattern of HBV subgenotype prevalence in Ribeirão Preto is more similar to that observed in cities in the South region of Brazil, which can be related with the historical context of settlement of the region. An intense European immigration to Brazil occurred in the XIX and XX centuries, which was stimulated by campaigns of the Brazilian government after the abolition of slavery to supply the necessity for labor force in the coffee plantations in São Paulo and colonize the Southern region of the country.25,26 Ribeirão Preto was one of the Southeast regions were the characteristic of the soil allowed the highest productivity of coffee production in the period of coffee expansion, which made this region an important place where European immigrants were brought when arriving in Brazil. These European immigrants were mostly Italians, who, particularly between the years 1890 and 1902 contributed to an explosive increase in the population of Ribeirão Preto, which went from 12,033 to 52,910 inhabitants, half (∼27,765) of them were of Italian origin.26
Currently HBV infection is under control among the Italian population as a result of several factors including the introduction of vaccination against HBV. However, until four decades ago HBV infection in Italy had a different epidemiological pattern with a medium endemic level (HBsAg>2% in general population) and wide geographical differences (South regions showing higher HBV prevalence), high proportion of HBeAg positivity, frequent detection of HDV infection, intra-familial transmission as the main way of HBV spread.27 Considering the past epidemiological HBV infection situation in Italy and the high prevalence of the HBV/D3 in this country the results found in our study confirm the conclusion of a previous study in Brazil about the great contribution of the Italians to the introduction and spread of HBV/D3 in Brazil,16,17 mainly in the regions colonized by these immigrants, as occurred in Ribeirão Preto. The closer relationship of HBV/D3 isolated in Ribeirão Preto with sequences from Italy can be observed in the phylogenetic tree that shows several branches with sequences from both regions (Fig. 2A).
As in most Brazilian regions, the studies done until now about HBV prevalence in Ribeirão Preto have shown low endemicity.28 In regions with this epidemiological characteristic the principal mode of HBV transmission is through risk behavior (unprotected sexual contact or sharing syringes with an HBV carrier) and through unsafe medical practices, whereas in high endemic regions most infections occur perinatally or during early childhood. Although Ribeirão Preto is an area of low HBV endemicity, household contact with an HBV carrier (excluding that with sexual contact) was identified as one of the most important risk factors for HBV transmission followed by sexual risk behavior in a previously conducted study, which suggests that intra-familial transmission is an important form of HBV spread in Ribeirão Preto.29 This hypothesis is reinforced now by the phylogenetic relationship showed by HBV sequences isolated from patients belonging to the same family (first-degree relatives). The pattern of intra-familial HBV transmission observed in this study was maternal transmission that can be vertical or horizontal. Some studies have suggested that persistent HBV infection in parents increase the frequency of intra-familial HBV infection, even more the case when the mother is infected.29–31
The role of intra-familial and vertical transmission of HBV was also suggested in two studies with conducted in South Brazil. In Chapecó, Santa Catarina state, having the mother and/or siblings carriers of HBV was the major risk factor to HBV infection identified among HBV chronically infected patients evaluated.17 In Paraná state the study of HBV prevalence showed a higher frequency of HBV markers (anti-HBc and HBsAg) among women belonging to families of Italian or German ancestry.32 Obviously, due to the introduction of HBV vaccination, currently intra-familial and vertical transmission is not the main means of HBV spread in Ribeirão Preto, but it is possible that it was in the past, as were in Italy some decades ago due to the maintenance of the immigrants life-style.33–35
Regarding the association of genotypes and subgenotypes with severe forms of the disease, although there are well-established associations between progression to cirrhosis and HCC in certain HBV genotypes, particularly related to genotype C found in Asia,1 no association between HBV genotype or subgenotype and the presence of severe disease liver was identified in this study. The cross-sectional design of the study should be taken into account, as well as the small number of subjects with cirrhosis and HCC.
ConclusionIn this study HBV D3 was most frequently detected subgenotype, followed by subgenotype A1. The HBV subgenotypes found this study are probably due to the mix of local population and to the migratory processes involved. The similarity of the HBV sequences encountered in first-degree relatives suggests the virus circulation in the domestic environment and eventual intra-familial transmission.
FundingThis work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP 08/11472-2.
Conflicts of interestThe authors declare no conflicts of interest.
