



Human adenoviruses comprise an important group of etiologic agents that are responsible for various diseases in adults and children, such as respiratory, ocular, gastroenteric, and urinary infections. In immunocompromised and organ-transplanted individuals, these agents can cause generalized infections. Rapid diagnostic methods for detecting these infectious agents are not widely available.
The aim of this work was to produce monoclonal and polyclonal anti-adenovirus antibodies to be used in a rapid diagnostic test for respiratory infections.
Adenovirus hexons were satisfactorily purified by ultracentrifugation and chromatography. After virus purification, anti-hexon monoclonal antibodies were produced and characterized, following classical methods. Antibodies were specific for adenoviruses 2, 3, 5, and 41. The proposed immunochromatographic test was standardized using colloidal gold.
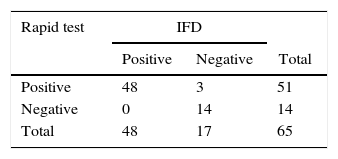
The standardization of the rapid test was sufficient to detect adenovirus antigens (in nasopharyngeal lavage samples) with sensitivity of 100% and specificity of 85% when compared to direct immunofluorescence.
The immunochromatographic assay prototype was sufficiently sensitive to detect B (3), C (2 and 5), and F (41) adenovirus samples. Although based on preliminary data, the test demonstrated the same performance as direct immunofluorescence, but with the advantage of being a point-of-care test. Further studies are still needed to confirm its effectiveness in clinical practice.
Human adenoviruses (HAdVs) are important agents that cause serious infections in children and immunocompromised patients. HAdVs were first detected in military personnel with acute febrile respiratory disease. Later, clinical manifestations such as gastroenteritis, cystitis, hepatitis, keratoconjunctivitis, meningoencephalitis, and myocarditis were also related to these viruses.1–10
The epidemiological studies of adenovirus infections have limitations due to the relatively high incidence of doubtful and inconclusive results obtained in the tests currently in use. Direct immunofluorescence (IFD) and Enzyme Linked ImmunonoSorbent Assay (ELISA) assays are techniques that are not very sensitive for the diagnosis of adenovirus infections, compared to cell culture methods and molecular diagnosis.3–5,10
Virus isolation in cell cultures is a sensitive method for adenovirus detection, but this method is costly and time-consuming, taking several days to perform the isolation. As some adenovirus serotypes are difficult to culture, in order to maximize sensitivity the virus should culture on at least two or three different cell lines.10 The turn-out of culture results are immediately provided to the clinician, and are impractical in studies with a large number of clinical samples.3,10
Molecular diagnosis by PCR (Polymerase Chain Reaction) is a relatively quick and sensitive tool, enabling direct laboratory diagnosis in the clinical sample.10 Depending on the DNA region amplified by PCR, it is possible to distinguish between species and serotypes.10 However, this technique still has a high cost and requires a specialized technical team to perform it, and it is therefore not very accessible.3,9,10
In this study, the purified adenovirus antigen was used in the production of monoclonal and polyclonal antibodies, which in turn were applied to a nitrocellulose membrane. The rapid test prototype consisted of anti-hexon polyclonal antibodies coupled to the glass fiber superimposed on high-flow nitrocellulose 180.
The combination of the 6D-1G monoclonal antibody on the test line (capture) with gold-conjugated rabbit polyclonal antibodies (fiber glass/detection) provided better specificity and sensitivity. When 6D-1G (concentration 1.2mg/mL) was tested with sucrose (3%) and trehalose (2.5%), there was successful detection of adenoviruses 2, 3, 5, and 41.
In the configuration of the proposed adenovirus detection test the monoclonal antibody was bound to a nitrocellulose membrane and the polyclonal antibody to a glass fiber membrane, and this proved to be a promising format in the standardization of an immunochromatographic assay for detecting all adenovirus serotypes.29
Material and methodsProduction of adenovirus hexons/purification stocks, electron microscopy and virus concentrationProduction of adenovirus stocks and purification, electron microscopy, and virus concentration were performed as previously described.11–19 The protein content was measured by bicinchoninic acid method.20
Production of polyclonal antibodiesImmunization of BALB/c miceVirus antigen was purified using cesium chloride gradient centrifugation and dialysis against 10mM HEPES buffer, pH 7.4, 10% glycerol and 1mM EDTA. Nine male BALB/c mice of approximately six weeks were immunized intraperitoneally (i.p.) as follows20: injection of 50μg adenovirus hexon protein (pure antigen) diluted in Freund's adjuvant. The booster dose was performed with i.p. injection of approximately 50μg antigen in Freund's incomplete adjuvant. The third dose was performed i.p. injection of approximately 50μg antigen in Freund's incomplete adjuvant. The fourth dose was administered with injection with 50μg antigen in Freund's incomplete adjuvant. Finally, spleen cells were used for the fusion.
Production/purification of polyclonal antibodies in rabbit was performed as previously described.20–22
Indirect ELISA of polyclonal antibodies from BALB/c miceSelection of positive mice, i.e., those that produced adenovirus antigen-specific antibodies, was based on the result of indirect ELISA test. The plate was coated with purified “adenovirus hexon” diluted in 0.05M carbonate/bicarbonate buffer, pH 9.6 (50μL/well), at a concentration of approximately 10μg/well. The plate was incubated overnight at 4°C and subsequently washed once with 0.05% PBST, and blocking solution (1% tryptone, 1% BSA, 3% sucrose (150μL/well)) in PBS for 1h at 37°C was then added. The plate was washed once with 0.05% PBST. Next, 50μL of sera were added to the ELISA plate, which was then incubated at 37°C for 1h. The negative control was PBS. The plate was washed 3 times with 0.05% PBST and then incubated with secondary antibody, horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) (Immunopure Pierce), diluted 1/10,000 in 0.05% PBST. Peroxidase activity was revealed by the addition of ELISA substrate TMB BD OptEIA. The reaction was stopped by adding 50μL/well of 3N H2SO4 and the plate read on a TP Reader Thermo Plate at a wavelength of 450nm.20
Monoclonal antibodies productionHybridoma technologyFusion was performed according to the protocol proposed by Dr. Leticia Barboza's group (Bacteriologia – Instituto Butantan). Myeloma cells from the Sp2/0-Ag14 line (ATCC CRL 1581-Köhler and Milstein, 1976) were purchased from the Rio de Janeiro Cell Bank (BCRJ). Cells were previously thawed, grown in pre-fusion medium (RPMI 1640 medium (Invitrogen) plus 10% NCTC (Invitrogen) and 10% FBS) and kept in an incubator at 37°C in 5% CO2.
For the preparation of hybridoma conditioned medium (feeder), mouse macrophages of the BALB/c strain were obtained by the intraperitoneal lavage procedure. Macrophages were obtained by introducing 5mL of RPMI 1640 medium (4°C) into the peritoneum. The medium was withdrawn with the macrophages and then centrifuged at 1200rpm for 5min. Finally, the cells were resuspended in RPMI 1640 medium with 10% NCTC and 10% FBS and incubated at 37°C in 5% CO2.
Initially, two 25-cm2 and three 75-cm2 flasks of SP20 cells were used in the assay. The cells were then counted in a Neubauer chamber and placed in a 5% CO2 incubator at 37°C, in approximately 5mL of pure DMEM. The BALB/c mice received the fourth booster dose of 50μg of purified “adenovirus” diluted in PBS three days prior to the fusion assay. The immunized animals were sacrificed and the spleen and lymph nodes removed. The spleen and lymph nodes were placed on a screen on top of a beaker and DMEM was injected with a needle and syringe (they swelled and burst easily and the cells fell through the screen). The tissue was macerated on the screen with the plunger of a glass syringe. The macerates were placed in a Falcon tube and centrifuged at 1200rpm for 5min. The supernatant was discarded and the splenocytes and lymph node cells were resuspended in 5mL of medium. The cells were then counted in a Neubauer chamber. SP2 cells and splenocytes (in 1:2 ratio) were mixed in a Falcon tube. Next, 10mL of pure DMEM were added and the tube was centrifuged at 1200rpm for 5min. The supernatant was discarded with the aid of a Pasteur pipette and the pellet was gently broken with 0.5mL of pure DMEM. Subsequently, 1mL of PEG was added with 50μL of DMSO for 1min, dropwise with slow stirring, in a beaker with water at 37°C, after which time the cells were allowed to remain another minute in the beaker. Subsequently, 2mL of pure DMEM were added dropwise with gentle shaking; an additional 20mL of pure DMEM were then added, and the cells were allowed to stand for 4min in water at 37°C. Thereafter, the tube with the cells was centrifuged at 1200rpm for 5min at room temperature. For lymph node cells, the pellet was resuspended in 10mL of complete medium: DMEM+3% HAT+15% FBS and plated (100μL) in a 96-well plate. For splenocytes, the pellet was resuspended in 30mL of complete medium: DMEM+3% HAT+15% FBS and plated (100μL) in three 96-well plates. Finally, the cells were incubated at 37°C, 10% CO2. The medium was changed every three days. After one week, 3% HAT was substituted with 2% HAT for another three weeks.21
Indirect ELISA of hybridoma supernatantSelection of positive hybridomas, i.e., those that produced adenovirus antigen-specific antibodies was performed by indirect ELISA. Prior to this, standardization of this ELISA test was performed using various concentrations of purified adenovirus antigen, HEK cell antigens (negative control), and commercial anti-adenovirus monoclonal antibody (positive control). This ELISA with cross-titration allowed a cut-off to be established so that truly positive hybridomas could be identified.
The plate was sensitized with purified adenovirus antigen diluted in 0.05M carbonate/bicarbonate buffer, pH 9.6 (50μL/well), at a concentration of approximately 0.5μg/well. In addition, all hybridoma supernatants were tested against HEK cell antigens (negative control) at a concentration of 0.0025ng/well. The plate was then incubated overnight at 4°C. Subsequently, it was washed once with 0.05% PBST and then blocking solution (1% tryptone, 1% BSA, 3% sucrose (150μL/well)) diluted in PBS for 1h at 37°C was added. Finally, it was washed once with 0.05% PBST, and then, 100μL of the hybridoma culture supernatant were incubated in the ELISA plate at 37°C for 1h. Non-immunized mouse serum was used as the negative control. Commercial anti-adenovirus monoclonal antibody (Merck/positive control) was incubated at a 1/3000 dilution. The plate was washed three times with 0.05% PBST and then incubated with goat anti-mouse IgG secondary antibody (H+L) conjugated to horseradish peroxidase (Immunopure Pierce) diluted 1/10,000 in PBS. Finally, peroxidase activity was revealed by the addition of ELISA substrate TMB BD OptEIA. The reaction was stopped by adding 50μL/well of 3N H2SO4 and the plate subsequently read in a spectrophotometer (TP Reader Thermo Plate) at 450nm.21
Limiting dilution of hybridomasHybridomas that had optical density close to or above the estimated cut-off (OD=0.548) – for purified adenovirus antigen – were subjected to limiting dilution. Hybridomas were detached from the flask and placed in a Falcon tube. The cells were then counted in a Neubauer chamber. The cell count of a 25-cm2 flask was around 3×105cells/mL. From this calculation, a sequential dilution in other Falcon tubes (with 10mL of culture medium) was done until approximately 50 cells in 10mL of medium were finally obtained. At the end, approximately 100μL were incubated in a 96-well cell culture plate and kept in an incubator at 37°C and 5% CO2.21
Monoclonal antibodies characterizationIndirect ELISA of purified monoclonal antibodiesThe plate was sensitized with adenovirus (serotypes 2, 3, 5, and 41) diluted with 0.05M carbonate/bicarbonate buffer, pH 9.6 (50μL/well), at a concentration of approximately 0.5μg/mL. In addition, all hybridoma supernatants were tested against parainfluenza virus (negative control) respiratory tract cell antigens at the same concentration, with overnight incubation at 4°C. Subsequently, the plate was washed once with 0.05% PBST and then incubated with blocking solution (1% tryptone, 1% BSA, 3% sucrose) in PBS (150μL/well) for 2h at 37°C. Wells were washed once with 0.05% PBST and then 100μL of purified monoclonal antibodies (5μg/mL) were added, followed by incubation at 37°C for 1h. The plate was incubated with anti-adenovirus monoclonal antibody (positive control) at 1/3000 dilution and washed six times with 0.05% PBST, and the secondary antibody, horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) (Immunopure Pierce), diluted 1/10,000 in PBS, was added, followed by incubation for 40min at 37°C. The reaction was stopped by adding 50μL/well of 3N H2SO4 and TMB BD OptEIA was added as substrate. The plate was read on a plate spectrophotometer (TP Reader Thermo Plate) at 450nm.21
ELISA for isotype was performed as previously described.21
Protocols for immunochromatographic testConjugation of colloidal gold with antibodyThe antibody(s) were resuspended (at a final concentration of 1mg/mL) in 0.2M NaCl–borate buffer pH 9.0, and then dialyzed against 2mM borate–NaCl buffer, pH 9.0 for 2h. The amount of antibody required to stabilize gold was determined: 100μL of colloidal gold (20nm, 1 OD or 0.01%, BBI) was added in aliquots containing 10μL of dilutions of dialyzed monoclonal antibody (in 2mM borate). After 10min, 11μL of 10% NaCl was added to each tube. The amount of antibody sufficient to stabilize the gold was the dilution in which the solution did not change color, i.e., insufficient amount of antibody changes the coloration of the solution from red to blue. Considering a dialyzed solution of antibodies at the approximate concentration of 1mg/mL, the volumes of 1–4μL were tested. Subsequently, the pH of the gold to be conjugated to pH 9.0 was adjusted with 0.1M K2O3 solution.
The colloidal gold was added to the stirring antibody, allowed to react for 30min at room temperature (10mL of gold colloidal in the amount of antibody determined in step 3). In order to stabilize the reaction, a 10% BSA solution (10% BSA in 0.01M borate buffer, pH 9.0) was mixed in sufficient volume to reach a final concentration of 1% and incubated for 30min. The solution was centrifuged at 15,000×g for 20min at 4°C and the supernatant discarded. The pellet was resuspended in 2mL of a 2% BSA in 0.01M borate buffer and centrifuged at 12,000×g for 20min at 4°C. Finally, the pellet was resuspended in 1mL of buffer (3% BSA, 3% sucrose, 0.01M sodium borate and 0.05% sodium azide) and stored at 4°C.23
Immunochromatographic assayEach paper type was treated separately before assembling the tape, as shown below: the sample pad(s) were immersed in buffer (20mM sodium borate buffer, 0.1% Triton X-100 and 0.1% sodium azide, pH 8.5) and stored in a vacuum chamber at room temperature for 2h or held at 56°C for 2h. Conjugate pad(s) (solid phases with conjugate) were soaked with 20μL of the pAb-gold solution. The fibers were then kept in an oven at 37°C for 2h. Nitrocellulose membranes (HiFlow Plus nitrocellulose 180 – Merck) were incubated with 0.1M Tris–HCl, pH 7.5, with 0.4% glycerol for 15min on a shaker. After immersion, the membranes were washed 1× with 10mM Na2HPO4 wash buffer pH 7.5 and then placed in the oven at 37°C for 18–20h for drying. Subsequently, 1.5μL 6D-1G monoclonal antibody (concentration 1.2mg/mL) diluted in 0.01M PBS pH 7.2 (test line) were applied. Then, 1.2mg/mL rabbit anti-IgG was applied after 0.5cm of the test line (control line). The membranes were then kept in a vacuum chamber for 2h. The strips were set for drying at room temperature overnight or in the vacuum chamber for 2h. The absorbing pad(s) (solid absorption phases) need no treatment. The diluent (0.01 PBS containing 0.1% Triton X-100 and 0.5% BSA) improved the migration rate of the sample (1:1 diluent/sample). For assembling the tests, the pretreated papers were overlapped according to Fig. 1.23
Ethics in Research Committee and control line (validation).")
The study was approved by the Ethics in Research Committee of the Universidade Federal de São Paulo, No. 0126/2011.
Results and discussionThe precise diagnosis of a case of adenovirus infection is important in order to avoid the spread of the disease, especially in closed institutions, such as nurseries, hospitals, clinics, schools and places with crowds of people1–5,8–10,22,24–26 as well as to avoid unnecessary use of medications.
Ideally, a diagnostic method for any infectious disease should be fast and inexpensive in the performance/outcome, have a good sensitivity/specificity ratio, and not require personnel with specialized training to perform it.9 Udeh et al. (2008) estimated that approximately $400 million per year of health resources could be saved with an accurate diagnosis of adenoviruses associated with conjunctivitis in the United States alone. In addition, 1.1 million cases of inappropriate use of antibiotics would be avoided.9,10
Viral isolation through cell culture remains the gold standard in the diagnosis of adenovirus because the isolation of the infectious agent is definitive. However, it is a time consuming technique, requires specialized personnel to perform, and has a high cost. The PCR technique is sensitive enough for detection, but it has a very high cost and requires highly qualified personnel for the diagnostic procedure.9,10
In the case of immunocompromised patients, rapid direct test (viral protein screening) has an advantage over indirect ELISA (serology) test since patients under these conditions do not have levels of antibodies high enough to be detected.5,10
Levent et al. (2009) evaluated an immunochromatographic assay for adenovirus (SAS adeno-test) and observed a sensitivity of 55% and specificity of 98.9% for samples from children with influenza-like illness.4 Romero-Gomez et al. (2014) analyzed the Adeno respiratory card letitest (Leti diagnostics Barcelona, Spain) in 224 samples of children under 15 who attended an emergency room. When compared to PCR, the rapid test showed sensitivity of 78% and specificity of 73%. In addition, the researchers speculated that the low specificity of the test indicated that it could be used in clinical decision making during the period of greatest adenovirus replicative activity.27
The antigen-capture immunochromatographic assay uses a strip consisting of three types of membranes (cellulose fiber, glass fiber, and nitrocellulose), mounted on a plastic support. In the strip, the cellulose fiber is located at the anterior and posterior ends, being interspersed by glass fiber and nitrocellulose. The test uses two antibodies that bind to different epitopes present in the molecule to be detected (analyte) (Fig. 1). One of the antibodies, labeled with a signal generator such as latex beads or colloidal gold, is called the detection antibody, while the second antibody is called the capture antibody. The detection antibody is placed in a dehydrated state on the glass fiber membrane, which will allow its instant dissolution upon contact with the aqueous medium containing the analyte. Under these conditions, the analyte binds to the antibody forming a complex, which migrates by capillarity on the strip until binding with the capture antibody, which was previously fixed on the nitrocellulose membrane. The cellulose fiber, located at the ends, provides the continuous flow of the complex. The color signal, generated by binding the complex to the capture antibody, is visualized as a line after 10–20min and can be analyzed qualitatively and/or quantitatively. For control of the reaction, a specific antibody to the detection antibody is placed on the nitrocellulose membrane on the same strip. The reaction is considered positive when both test and control lines show the characteristic color after the reaction time.2,4,6,8,25–27
In our study, the processes of adenovirus antigen production and chromatography resulted in good purification of the material.13,14,18,20,22,28 Elution fractions of the hexon protein were found at the same concentrations as previously described.13–17,20,22 After immunoblotting, it was confirmed that the polyclonal serum of BALB/c mice contained specific antibodies against adenovirus hexon as previously described.15,20,22 The hexon immunization protocol used by Hongling et al. (2009) was satisfactory, since excellent humoral response was obtained in BALB/c mice and rabbits. Three hybridomas (6D-1G, 6D-4E, and 6D-7C) were maintained in culture and the antibodies produced were able to recognize cultured adenovirus (prototypes) 2, 3, 5, and 41 (Fig. 2). The cutoff value to determine the reactivity of different monoclonal antibodies was established by testing seronegative samples for adenovirus antibodies and multiplying the medium value by 2.1 (95% upper limit of variability). All hybridomas showed isotype IgG1 (data not shown).
.")
In this study, when 6D-1G antibody (concentration 1.2mg/mL with sucrose 3% and trehalose 2.5%) was tested, detection of adenovirus 2, 3, 5, and 41 was successful (Fig. 3). The fiberglass containing polyclonal antibodies (concentration 1.2mg/mL) was superimposed on the nitrocellulose membrane (HiFlow Plus nitrocellulose 180). The diluent (0.01M PBS containing 0.1% Triton X-100 and 0.5% BSA) improved the migration rate of the sample. This format was promising since 100% sensitivity and 85% specificity was achieved in the detection of adenovirus in samples of pediatric patients already analyzed by IFD (Table 1). New assays with a larger number of samples are still needed to confirm this performance. Negative samples can be targeted for cell culture testing and PCR.
 2, 3, 5, and 41 was successful.")
This first work aimed to describe in more detail the preparation of a prototype and did not intend to evaluate the test in its final format, although the preliminary results were quite encouraging. Once standardized in greater detail, this rapid assay prototype will have applicability in the accurate and rapid diagnosis of respiratory infections at the emergency room. Its use will bring benefits when there is suspicion of outbreaks, allowing the physician to prescribe immediate and appropriate treatment for the disease.
FundingFapesp-Fundação de amparo a pesquisa do estado de São Paulo/SP/Brasil. Process number 2011/50100-6.
Conflicts of interestThe authors declare no conflicts of interest.
To Dr. Carlos Taborda (Departamento de Microbiologia, Instituto de Ciências Biológicas) for allowing use of laboratory equipment. To Dr. Roxane Piazza (Departamento de Bacteriologia, Instituto Butantan) for providing laboratory facilities and protocols. To Dr. Pascal Fender and Daphna Fenel (Laboratoire de Microscopie Electronique Structurale – Grenoble/France) for assistance with transmission electron microscopy. Dr. A. Leyva provided English translation and editing.
