






Antiviral drug resistance is the most important factor contributing to treatment failure using nucleos(t)ide analogs such as lamivudine for chronic infection with hepatitis B virus (HBV). Development of a system supporting efficient replication of clinically resistant HBV strains is imperative, and new antiviral drugs are needed urgently to prevent selection of drug-resistant HBV mutants. A novel fluorinated cytidine analog, NCC (N-cyclopropyl-4′-azido-2′-deoxy-2′-fluoro-β-d-cytidine), was recently shown to strongly inhibit human HBV in vitro and in vivo. This study was designed to evaluate the antiviral activity of NCC against lamivudine-resistant HBV. We generated a stable cell line encoding the major pattern of lamivudine-resistant mutations rtL180M/M204V and designated it “HepG2.RL1”. Immuno-transmission electron microscopic examination and enzyme-linked immunosorbent assay were used to detect secretion of HBV-specific particles and antigens. Quantification of extracellular DNA and intracellular DNA of HepG2.RL1 cells by quantitative real-time polymerase chain reaction revealed >625-fold and >5556-fold increases in the 50% inhibitory concentration of lamivudine, respectively, compared with that for the wild-type virus. The results showed that NCC inhibited DNA replication and HBeAg production in wild-type or lamivudine-resistant HBV in a dose-dependent manner. In conclusion, screening for antiviral compounds active against lamivudine-resistant HBV can be carried out with relative ease using hepG2.RL1 cells. NCC is a potential antiviral agent against wild-type HBV and clinical lamivudine-resistant HBV and deserves evaluation for the treatment of HBV infection.
Hepatitis B virus (HBV) infection is a global health problem, with more than 240 million individuals chronically infected worldwide and about 30 million in China.1 Chronic infection with HBV is closely associated with liver cirrhosis and hepatocellular carcinoma, and carries high mortality.2–4 In recent years, on account of vaccines as well as treatment using interferon or nucleos(t)ide analogs,5 the prognosis of hepatitis B has improved significantly. However, HBV infection-related diseases remain a concern.
Covalently closed circular HBV DNA is a major determinant of persistence of the HBV genome in infected cells, and necessitates long-term antiviral treatment in those infected individuals.6,7 Unfortunately, prolonged administration of nucleos(t)ide analogs can result in the emergence of drug-resistant strains, especially if nucleos(t)ide analogs with low genetic barriers to resistance are used as monotherapies.8–10 For lamivudine, the prevalence of resistance rises rapidly, reaching 80% after five years of monotherapy.11 The pattern of rtL180M/M204V is the most common lamivudine-resistant HBV variant, with a prevalence of 60%.12 Emergence of lamivudine resistance leads to acute exacerbation or treatment failure of hepatitis and decompensation of liver function.13,14 Thus, novel drugs for the treatment of HBV, especially for rescue therapy in drug-resistant patients, are urgently needed.
NCC, N-cyclopropyl-4′-azido-2′-deoxy-2′-fluoro-β-d-cytidine, is a novel fluorinated cytidine analog. Recent studies have demonstrated that NCC is an efficient inhibitor of wild-type HBV in HepG2.2.15 and HepG.CW cells.15 However, the activity of NCC on a lamivudine-resistant HBV mutant remains unclear. The commonly used experimental approach is transient transfection of human hepatoma cells.16 However, in this way, the HBV genome is not integrated into host cells and only temporarily produces HBV particles, and the level of HBV expression is unstable. Here, a novel cell line was generated through stable transfection of an HBV genome (1.3-unit length) with lamivudine-resistant mutations of rtL180M and rtM204V into hepatoma HepG2 cells.
In this study, HepG2.2.15 cells and the newly generated cell line were used as experimental models to investigate the anti-HBV activity of NCC. Our results demonstrated that NCC possesses significant inhibitory activity against lamivudine-resistant HBV.
Materials and methodsPatient specimenThe patient was a 49-year-old Chinese female with hepatitis B e antigen-positive chronic HBV infection. Initially, she received lamivudine monotherapy (100mg per day, p.o.) and had serum levels of HBV DNA of 8.23log10copies/mL. After receiving lamivudine for five months, the viral load decreased to 4.39log10copies/mL, and then became undetectable after an additional three months. Unfortunately, HBV DNA rebounded to 6.86log10copies/mL and rose continuously in the next two months. At this point, signature mutations of lamivudine resistance (rtL180M/M204V) were detected by DNA sequencing of the reverse transcriptase domain of circulating HBV, and the genotype was of clone B (Invitrogen, Shanghai, China). Blood samples were collected after viral breakthrough.
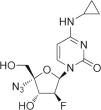
CompoundsLamivudine was obtained from GlaxoSmithKline (Suzhou, China). Adefovir (GlaxoSmithKline, Tianjin, China) was used as a positive control for inhibition of the lamivudine-resistant HBV strain. NCC (Fig. 1) with 98.5% purity (as determined by high-performance liquid chromatography) was designed and synthesized in our laboratory (The College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou). Stock solutions (10mM) of the three compounds were dissolved in phosphate-buffered saline.
Generation of plasmids expressing lamivudine-resistant HBV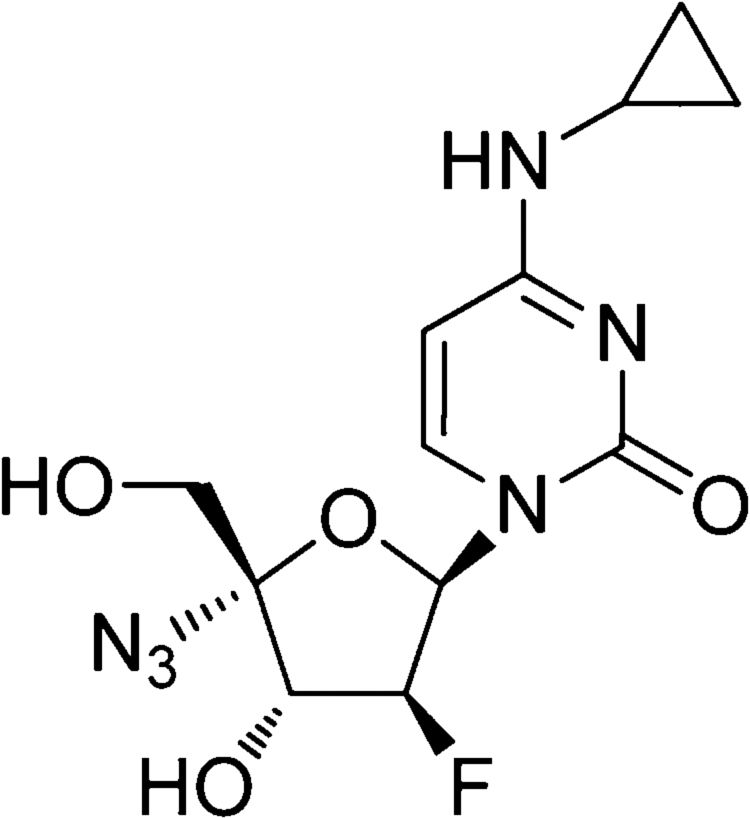
HBV DNA was extracted using a Viral DNA Extraction kit (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer's instructions. Briefly, patient serum was digested by OB protease and Buffer BL with linear acrylamide and RNase A, and then incubated sample at 65°C for 10min, followed by lysate with absolute ethanol (room temperature, 96–100%). The lysate was transferred into a HIBind DNA mini column and cleared of liquid by centrifugation at 8000×g for 1min to bind DNA. The HIBind DNA mini column was washed by Buffer HB and DNA wash Buffer and cleared of liquid by centrifugation at 15,000×g for 2min to dry the column. After incubated the column by preheated Elution Buffer for 5min at room temperature, DNA was eluted by centrifugation at 8000×g for 1min. Polymerase chain reaction (PCR) amplification of the full-length HBV genome was performed according to the methods of Günther et al.,17 with the primers p1 and p2 (shown in Table 1). Cycling parameters: pre denaturation 94°C for 5min; 94°C for 40s, 60°C for 78s, 68°C for 3min, the extension time plus 1min after every 10 cycles, 30 cycles total; final additional extension at 72°C for 10min; hold at 4°C. Amplicons were cloned into pMD18-T (TaKaRa Bio, Dalian, China) to generate the T-HBV recombinant for sequencing the reverse transcriptase domain. Results showed that the cloned HBV genome harbored rtL180M/M204V mutations, and that the genotype was clone B.
Primer sequence for generation of pcDNA3.1-1.3 HBV recombinant plasmid.
| Name | Sequence (5′–3′) and restriction sites | Fragment size | Location in HBV genome | |
|---|---|---|---|---|
| HBV genome | P1: | GGCACTAGTGAGCTCTTCTTTTTCACCTCTGCCTAATCA | 3215bp | 1–3215 |
| P2: | TAAGCGGCCGCGAGCTCTTCAAAAAGTTGCATGGTGCTGG | |||
| A | A1: | GCAAGCTTGGACGACCCGTCTCGGGGCCGTTT Hind III | 370bp | 2846–3215 |
| A2: | GGCAGAGGTCAAAAAGTTGCATGGTGCTGGTGAACA | |||
| B | B1: | ACCAGCACCATGCAACTTTTTCACCTCTGCCTAATCATC | 1393bp | 1–1393 |
| B2: | GTTGTGGAATTCCACTGCATGGCCTGAGGA EcoR I | |||
| C | C1: | GCAGTGGAATTCCACAACATTCCACCAAGCTCT EcoR I | 1822bp | 1394–3215 |
| C2: | GGCAGAGGTCAAAAAGTTGCATGGTGCTGGTGAACA | |||
| D | D1: | ACCAGCACCATGCAACTTTTTCACCTCTGCCTAATCATC | 495bp | 1–495 |
| D2: | TATGCGGCCGCAGGGGCATTTGGTGGTCTGTAA Not I | |||
| A+B | A1: | GCAAGCTTGGACGACCCGTCTCGGGGCCGTTT Hind III | 1763bp | 2846–1393 |
| B2: | GTTGTGGAATTCCACTGCATGGCCTGAGGA EcoR I | |||
| C+D | C1: | GCAGTGGAATTCCACAACATTCCACCAAGCTCT EcoR I | 2317bp | 1394–495 |
| D2: | TATGCGGCCGCAGGGGCATTTGGTGGTCTGTAA Not I | |||
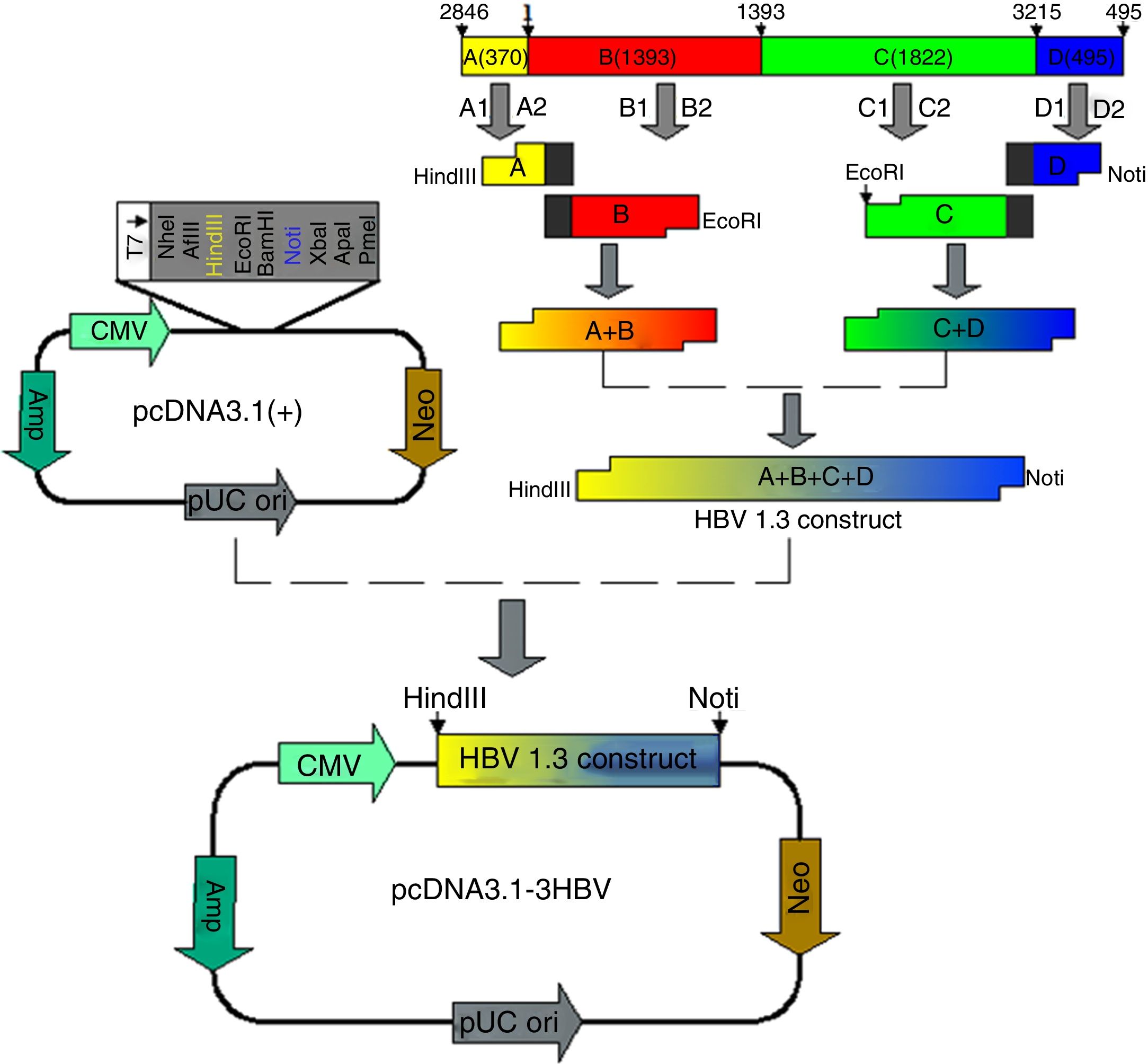
HBV DNA is circular and partially overlapped, and its genome comprises four open reading frames named S, C, P, and X. According to this feature of HBV genome structure, we designed and amplified four fragments designated A, B, C, and D, and constructed a recombinant plasmid harboring 1.3 units length of HBV genome (4080bp) to ensure efficient HBV replication and expression. Fragment A is located from nt 2846 to nt 3215 of HBV genome, fragment B is located from nt 1 to nt 1393 of HBV genome, fragment C is located from nt 1394 to nt 3215 of HBV genome, and fragment D is located from nt 1 to nt 495 of HBV genome (A+B+C+D=4080bp) (Table 1). T-HBV recombinant was used as a template for amplification of A, B, C, and D gene fragments using the corresponding primers (shown in Table 1) with the following reaction procedure: 94°C for 4min, then 30 cycles of 94°C for 30s, 58°C for 30s, 72°C for 1min, and 72°C for 6min. The A+B and C+D gene fragments were obtained by recombinant PCR with the primers (shown in Table 1) and A, B and C, D gene fragments, respectively, as templates, and the amplification condition was the same as above. Fragment A+B was then digested with HindIII and EcoRI, and fragment C+D was digested with EcoRI and NotI. A 1.3-fold-over- length HBV genome containing fragments A+B and C+D was then ligated into HindIII and NotI cleaved pcDNA 3.1 plasmids by T4 DNA Ligase at 16°C for 16h, resulting in pcDNA3.1-1.3HBV.
Generation of stable cell line and detection of HBV antigens and HBV DNAThe human HepG2 was obtained from the China Center for Typical Culture Collection (Wuhan, China). Cells were cultured in DMEM (Dulbecco's modified Eagle's medium, Gibco, New York, NY, USA). HepG2 cells were transfected with pcDNA3.1-1.3HBV encoding mutant HBV using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Two days after transfection, cells were selected in medium supplemented with 600μg/mL G418 (Sigma–Aldrich, Saint Louis, MO, USA) for three weeks and renewed every three days. The G418-resistant clone was transferred to a T25 culture flask and maintained in the same Dulbecco's modified Eagle's medium with G418 at 300μg/mL. The novel stable cell line was designated “HepG2.RL1 cell.” The surface antigen of HBV (HBsAg) and e antigen of HBV (HBeAg) in cell culture supernatants were detected using an enzyme-linked immunosorbent assay (ELISA) kit (Kehua Bio-engineering, Shanghai, China), and intracellular HBV DNA was extracted from the cells by Tissue viral DNA Extraction Kit (Omega Bio-Tek, USA) according to the manufacturer's instructions, respectively. The amount of intracellular and extracellular HBV DNA were determined by quantitative real-time PCR (qPCR) based on the TaqMan technology and performed in a Light-Cycler (Roche, Mannheim, Germany) with the HBV Fluorescent Quantitative PCR Detection Kit (Qiagen CO., Ltd. Shenzhen, China) following the manufacturer's protocol. After an initial denaturation at 95°C for 90s, 40 cycles of denaturation at 94°C for 5s and annealing/extension at 56°C for 45s were conducted.
Examination of HBV-specific particlesCell medium (30mL) was collected and centrifuged at 3000×g for 30min at room temperature. The supernatant was filtered through a 0.22μm filter membrane. HBsAg (+) healthy blood serum (4mL) was diluted 1:5 with PBS and also filtered through a 0.22μm filter membrane. Subsequently, filtrates were mixed at 1:1 and incubated overnight at 4°C, then was centrifuged and resuspended in culture medium. Suspensions were placed on a copper network supported by carbon membranes. They were examined under immuno-transmission electron microscopy (ITEM) after negative staining with 2% phosphotungstic acid for observation of virus particles.
Cell culture and cytotoxicity assayHepG2.2.15 cell line (wild-type HBV in the stable HBV-producing) was obtained from the Wuhan Institute of Virology (Wuhan, China) and maintained in the same manner as HepG2 cells except that 200μg/mL G418 was added to the culture medium in our laboratory. Compound cytotoxicity was determined by the MTT assay on HepG2.2.15 cells and HepG2.RL1 cells.18 Cells were treated with lamivudine, adefovir, and NCC at various concentrations (62.5, 125, 250, 500, and 1000μM).
Drug susceptibility assays19HepG2.2.15 cells and HepG2.RL1 cells were seeded in 24-well culture plates at 3×104 cells per well and cultured for 24h. Further, lamivudine and adefovir at various concentrations and NCC (0.04, 0.2, 1, 5μM) were added to cells for six days. On day 6, HBV DNA was quantified as described previously by qPCR to assess the inhibitory efficiency of drugs on viral replication. HBsAg and HBeAg production were assessed by ELISA. The 50% effective concentration (EC50) was calculated by regression analyses. Fold resistance was calculated as the ratio of mutant EC50 to wild-type EC50.
Statistical analysesStatistical analyses were performed using SPSS v17.0 (IMB, Armonk, NY, USA). Results are the mean±SD. One-way ANOVA was used to determine the statistical significance of differences between groups. p<0.05 was considered significant.
ResultsObtaintion of HBV fragments and the pcDNA3.1-1.3HBV recombinant plasmidAs shown in Fig. 2A, the full-length HBV genome was extracted from the patient serum and amplified by PCR. The amplicon was cloned into pMD18-T, and used as a template for amplification of A, B, C, and D gene fragments. The PCR products were purified with Gel Extraction Kit (Omega Bio-Tek, USA) from an agarose gel, with a single strip (Fig. 2B). The A+B and C+D gene fragments were obtained by recombinant PCR, and digested with HindIII and EcoRI, EcoRI and NotI, respectively, then purified with Gel Extraction Kit (Fig. 2C). The gel purification of double-enzyme A+B and C+D gene fragments were ligated into the Hind III and Not I-digested pcDNA 3.1 plasmid (Fig. 2D), producing a recombinant clone termed pcDNA3.1-1.3HBV (Fig. 3). For restriction double-enzyme digestion by Hind III and Not I, the recombinant plasmid pcDNA3.1-1.3HBV was separated into two bands with pcDNA 3.1 plasmid (5428bp) and A+B+C+D (4080bp) (Fig. 2E), as well as digestion by Hind III, the 9.5kb linear plasmid pcDNA3.1-1.3HBV was shown (Fig. 2F). The full sequence of cloned pcDNA3.1-1.3HBV was uploaded as a supplementary file (Doc.1).
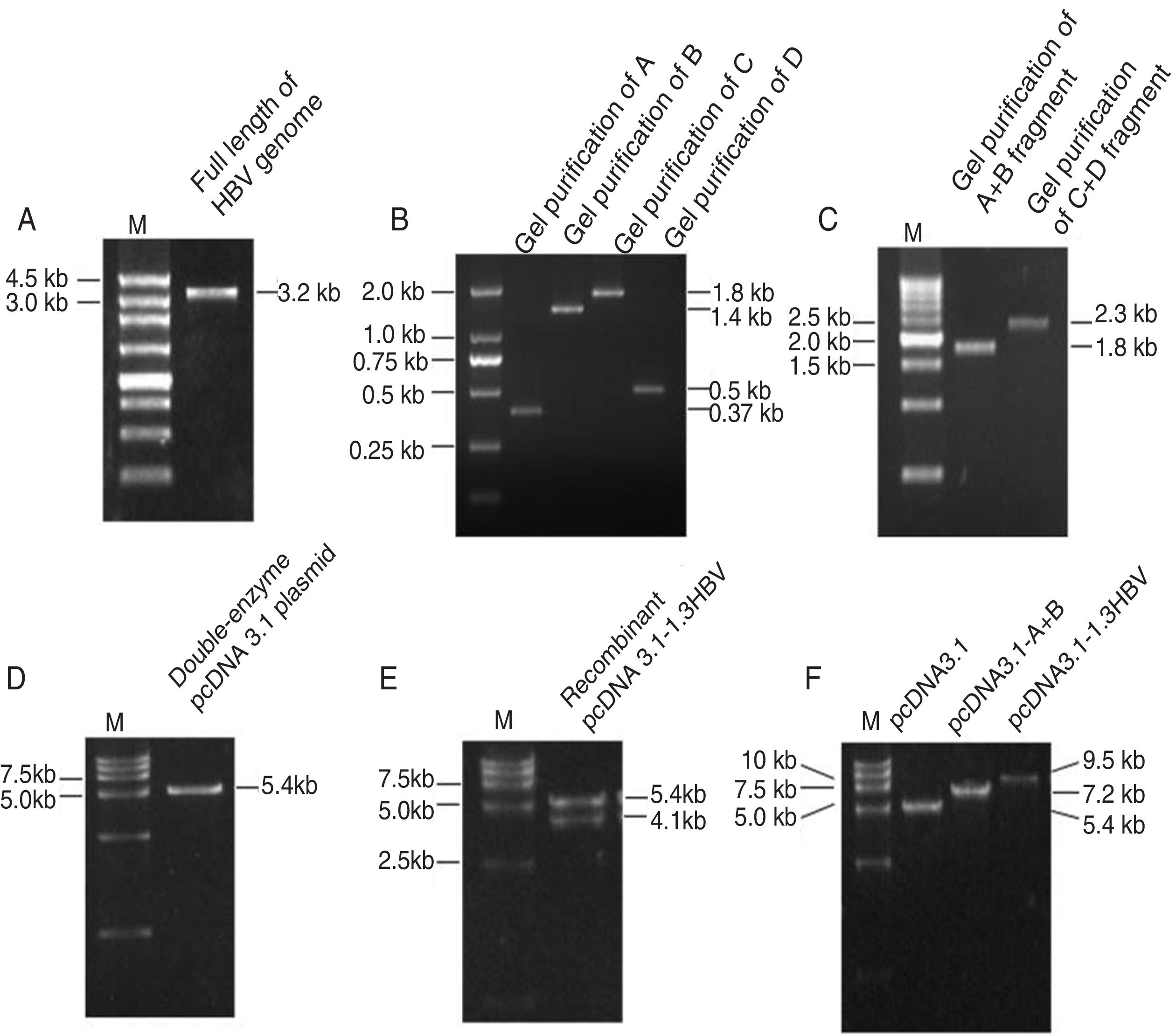
The PCR products of agarose gel electrophoresis analysis.
(A) Full-length HBV genome amplified by PCR. Lane M: 250bp DNA Ladder Marker; Lane 1: the full length HBV genome (3215bp). (B) Gel purification of A, B, C and D PCR fragments. Lane M: DL2000 DNA Marker; Lane 1, 2, 3,4: gel purification of A (370bp), B (1393) bp, C (1822bp), and D (495bp) PCR fragment, respectively. (C) Gel purification of A+B and C+D restriction enzyme digestion fragments. Lane M: 500bp DNA Ladder Marker; Lane 1: gel purification of A+B fragment digested by Hind III and EcoR I; Lane 2: gel purification of C+D fragment digested by EcoR I and Not I. (D) Gel purification of double-enzyme digested pcDNA 3.1 plasmid. Lane M: DL15000 DNA Marker; Lane 1: gel purification of double-enzyme pcDNA 3.1 plasmid (5428bp) digested by Hind III and Not I. (E) Restriction double-enzyme digestion identification of recombinant plasmid pcDNA3.1-1.3HBV by Hind III and Not I. Lane M: DL15000 DNA Marker; Lane 1: digestion of recombinant plasmid pcDNA3.1-1.3HBV by Hind III and Not I as pcDNA 3.1 plasmid (5428bp) and A+B+C+D (4080bp). (F) Enzyme digestion identification of recombinant plasmid pcDNA3.1-1.3HBV by Hind III. Lane M: DL15000 DNA Marker; Lane 1: digestion of plasmid pcDNA3.1by Hind III; Lane 2: digestion of recombinant plasmid pcDNA3.1-A+B by Hind III; Lane 3: digestion of recombinant plasmid pCDNA3.1-1.3HBV by Hind III.
To further characterize HepG2.RL1 cells, the supernatants and cells of each generation were collected for 100 continuous passages. HBeAg and HBsAg were secreted continuously at a stable level (Fig. 4A). The level of HBeAg produced by HepG2.RL1 cells was higher than the level of HBsAg under identical conditions. Amounts of viral DNA increased markedly at the 20th generation during the culture period. At the 10th and 100th generations, HepG2.RL1 cells produced 5.67 and 8.08log10copies/mL of intracellular DNA, as well as 4.51 and 5.61log10copies/mL of extracellular DNA, respectively (Fig. 4B).
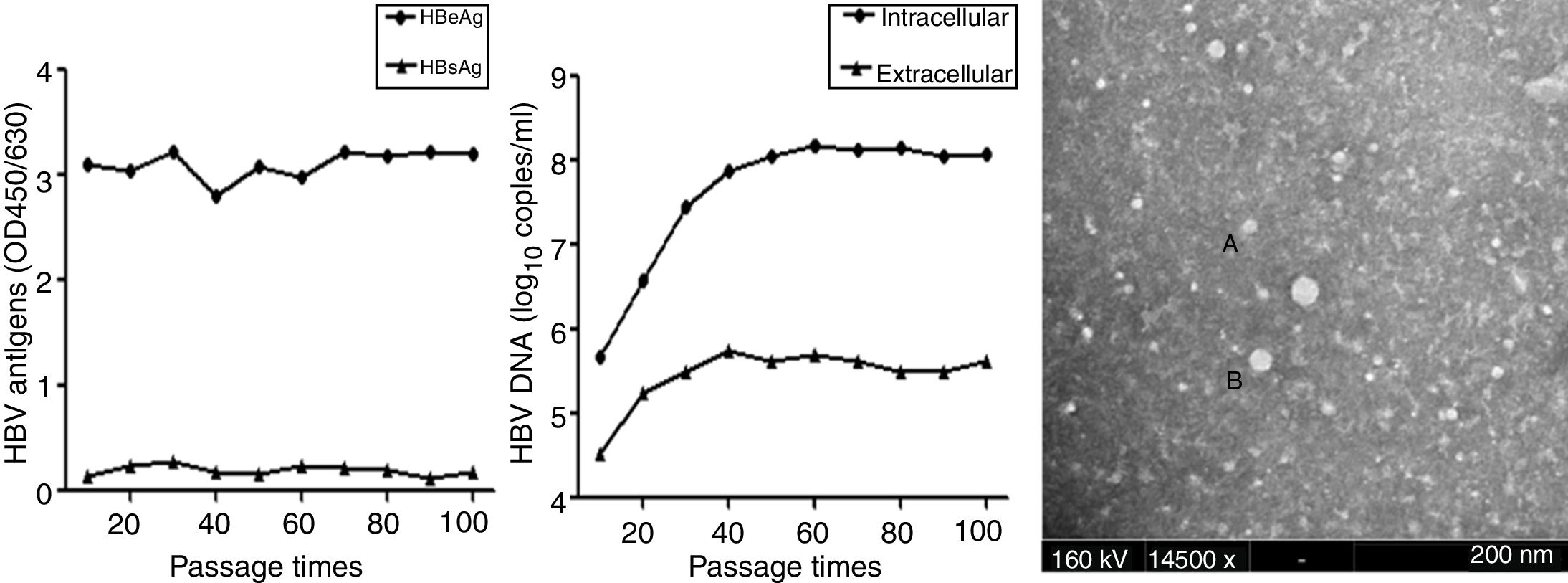
Time-course of secretion of viral antigens and HBV DNA, and electron micrograph of HBV particles in the HepG2.RL1 cell line
(A) Levels of HBeAg and HBsAg in the culture supernatants of the HepG2.RL1 cell line according to generation. OD450/630, optical density at 450nm and 630nm. (B) Quantity of intracellular and extracellular DNA in HBV according to generation. (C) Shown here is a typical complex that includes some spherical HBsAg particles (diameter, 22nm) (A) and a few Dane-like particles (42nm) (B) (magnification 14,500×; bar: 200nm; accelerating voltage: 160kV).
Samples containing medium of the HepG2.RL1 cells were visualized by ITEM analyses. Immune complexes were obtained from samples that had been immunoprecipitated with antiserum directed against HBsAg. Some of these immune complexes had several Dane-like particles of size ∼42nm and a few spherical HBsAg particles (∼22nm) (Fig. 4C). However, when samples containing medium of HepG2 cells were examined, virus-associated particles were not observed.
Effects of compounds on cell viabilityUpon exposure to various concentrations of NCC for six days, the results from the MTT assay showed that NCC inhibited the growth of HepG2.2.15 cells and HepG2.RL1 cells with CC50 of 725.81±23.34μM and 782.38±12.17μM, respectively, indicating almost the same extent of toxicity on the two cell lines, whereas CC50 for adefovir was 259.81±13.49μM and 290.57±21.31μM in HepG2.2.15 cells and HepG2.RL1 cells, respectively.
Drug susceptibility assaysThe HBsAg level from HepG2.RL1 was very low even in the drug-free group (data not shown) and hence the evaluation was based only on HBeAg. The dose-response relationship showed that lamivudine did not inhibit HBeAg secretion at the highest concentration (100μM) in HepG2.RL1 cells, whereas wild-type HBV showed considerable inhibition (Fig. 5A). However, adefovir treatment dose-dependently reduced the secretion of HBeAg in wild-type and lamivudine-resistant HBV (Fig. 5B).
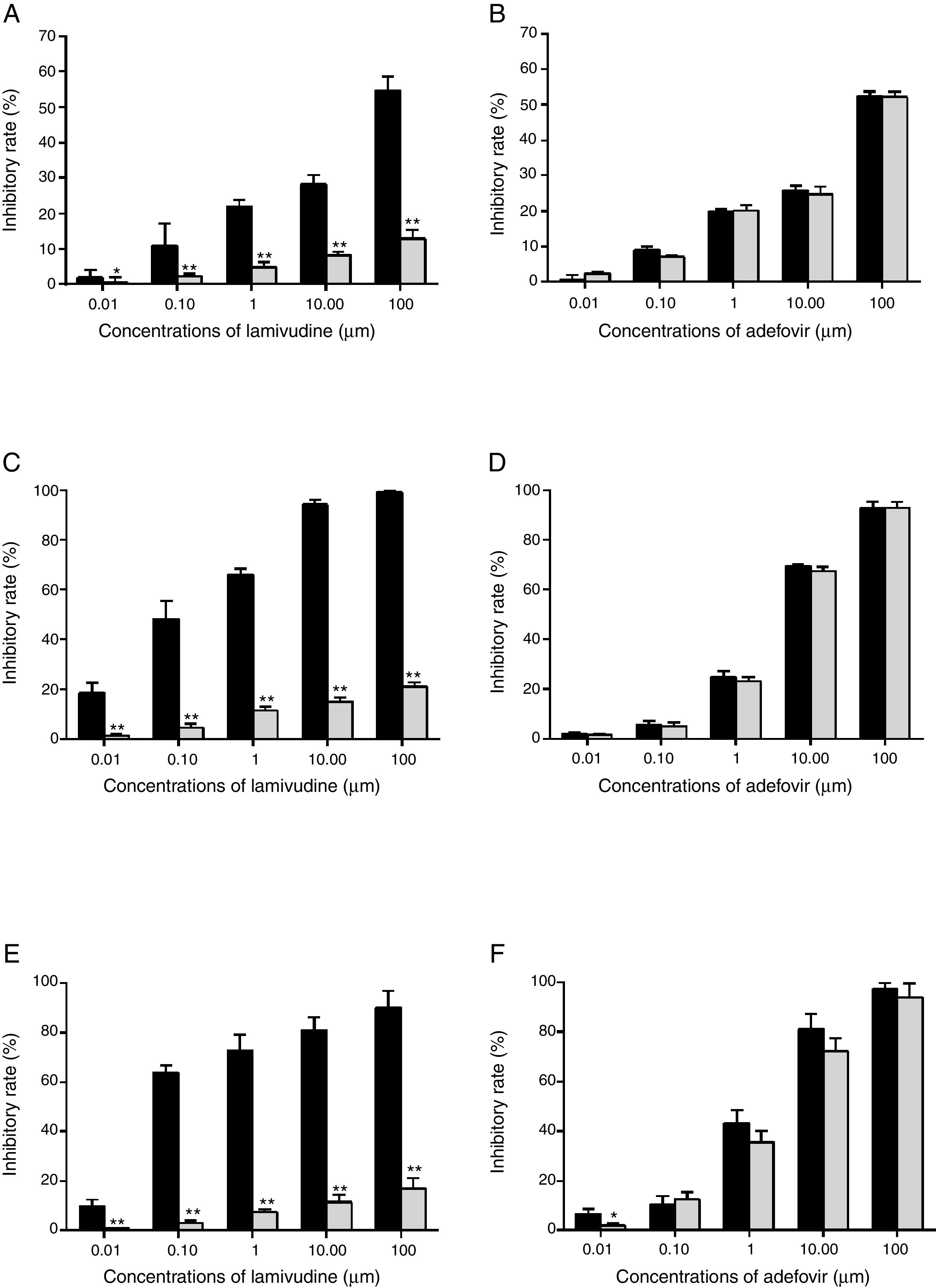
Drugs susceptibility to antigens secretion and viral replication.
HepG2.2.15 cells and HepG2.RL1 cells were treated with lamivudine and adefovir for six days. Cell-culture supernatants were assayed using a specific ELISA kit. Viral DNA was extracted from the culture medium and cells, and then quantified by qPCR using a commercial kit. Experiments were done from wild-type and lamivudine-resistant HBV isolates, on the inhibitory effects of lamivudine (A) and adefovir (B) on HBeAg production; inhibitory efficiency of lamivudine (C) and adefovir (D) on extracellular production of HBV DNA; inhibitory efficiency of lamivudine (E) and adefovir (F) on intracellular production of HBV DNA. Mean values of inhibition from three independent measurements are shown as bars with standard-error values indicated at the top. **p<0.01, *p<0.05, compared with the no-drug control group. Black bar: wild-type HBV; gray bar: lamivudine-resistant HBV with rtL180M/M204V mutations.
Viral DNA was extracted and subjected to qPCR analyses at the end of treatment. In terms of inhibitory potency on extracellular DNA replication, EC50 of lamivudine from wild-type HBV and HepG2.RL1 cells were 0.16±0.06μM and >100μM, respectively (Fig. 5C), which indicates a >625-fold-resistance. Based on the dose-response histogram, adefovir inhibited the replication of mutant HBV almost as efficiently as it did with wild-type HBV, with EC50 of 4.18±0.18μM and 3.69±0.50μM, respectively (Fig. 5D). With respect to intracellular DNA, lamivudine was not active on the rtL180M/M204V double mutation carrying HepG2.RL1 cells (EC50>100μM) compared to the wild-type HBV strain (EC50=0.018±0.01μM), which represents a decreased susceptibility of more than 5556-fold (Fig. 5E). Adefovir inhibited replication of intracellular DNA in HepG2.RL1 cells, consistent with that seen in wild-type HBV, with EC50 of 2.24±0.11μM and 1.36±0.08μM, respectively, both in a dose-dependent manner (Fig. 5F).
Effect of NCC on antigen production and viral replication in the two cell linesInhibitory efficiency of antiviral agents was assessed only with HBeAg. NCC dose-dependently inhibited HBeAg production by HepG2.2.15 cells and HepG2.RL1 cells (Fig. 6A). Consistent with the inhibitory effect on HBeAg production, NCC inhibited the replication of wild-type HBV and resistant HBV intracellular DNA in a dose-dependent manner. Importantly, there had no significant difference of the inhibitory rate between HepG2.2.15 cells and HepG2.RL1 cells (Fig. 6B), and EC50s for the inhibition of replication of intracellular HBV DNA by NCC were 2.23±0.79μM and 0.84±0.12μM, respectively. Moreover, NCC exposure produced dose-dependent inhibition of the replication of extracellular HBV DNA (Fig. 6C), with EC50s of 0.14±0.07μM and 0.46±0.09μM, respectively, for the HepG2.2.15 cells and HepG2.RL1 cells.
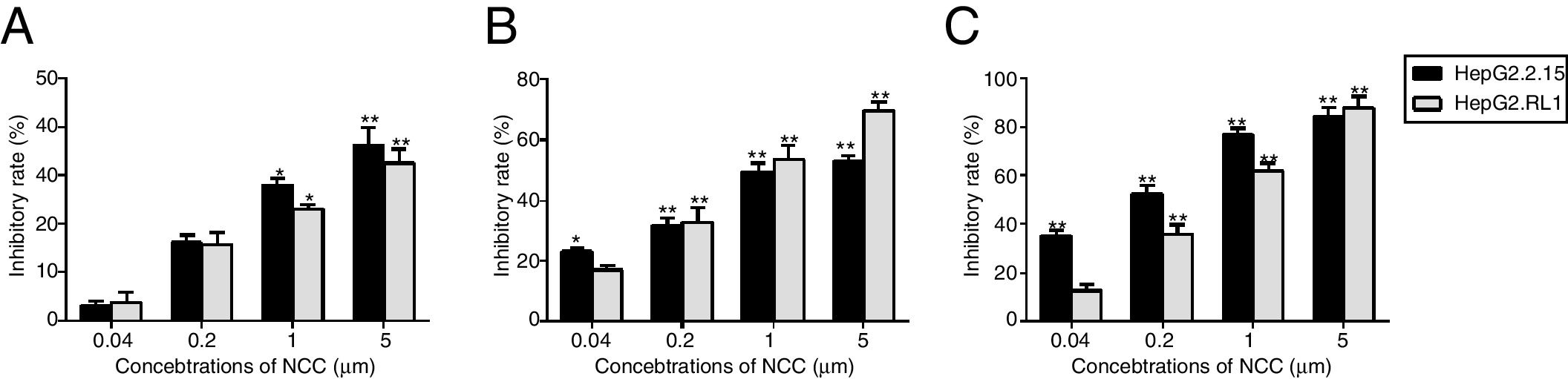
Effect of NCC on antigen secretion and viral replication.
HepG2.2.15 cells and HepG2.RL1 cells were treated with the indicated concentrations of NCC for 6 days. HBeAg was tested using a specific ELISA kit (A). The intracellular DNA (B) and extracellular DNA (C) was quantified by qPCR using a commercial kit. Experiments on the inhibitory effects of NCC on HBeAg production and replication of HBV DNA from HepG2.2.15 cells (black bar) and HepG2.RL1 cells (gray bar) were performed in triplicate. Mean values of inhibition from three independent measurements are shown as bars with standard-error values indicated at the top. **p<0.01, *p<0.05, compared with the no-drug control group.
In recent years, lamivudine has still remained widely used in the treatment of chronic hepatitis B either singly or in combination with other agents, mostly owing to its low cost.20,21 The prolonged use of lamivudine is associated with emergence of principal mutations in the tyrosine-methionine-aspartate-aspartate (YMDD) motif within the C domain of HBV polymerase, which are the substitutions of valine, isoleucine, or (rarely) serine for methionine (rtM204I/V/S).22,23 The rtM204I substitution has been detected in isolation, but rtM204V and rtM204S are found only in association with other changes, which are at domain A (L80V/I, L82M, L91I)24 and B (rtV173L, rtL180M).25,26 These additional compensatory mutations may stem from the natural evolution of the dynamics of quasispecies of HBV.27 HBV genotypes have distinct geographical distributions and correlate with different clinical outcomes. Previous reports indicated that genotypes B and C are predominant in China.28 Therefore, serial plasmids containing a specific HBV genotype (e.g., genotype B) and lamivudine-resistant have more practical clinical value.
In the HBV genome, the surface gene is overlapped completely by the polymerase gene.29 Hence, mutations in the reverse transcriptase domain associated with lamivudine-resistance might produce sequence/structural changes in the surface gene and its envelope protein that result in reduced antigenicity of the protein in the surface antigen or provoke stop codons in the surface gene, and may even affect the replication and infectivity of the virus.30–32 It has been suggested that ELISA cannot detect the I195M HBsAg protein efficiently.33 Thus, HepG2.RL1 cells secreted HBsAg at a lower level than HBeAg during the previous 100 generations, and the inhibitory efficiency of antiviral agents in this study was assessed based only on HBeAg production.
The impact of the lamivudine-resistant mutants on the observed efficacy of the drugs can best be described by the corresponding fold-resistance values in EC50in vitro. For lamivudine, treatment failure is associated with >100-fold increase in EC50. In our drug susceptibility testing, the fold changes in lamivudine activity against extracellular DNA and intracellular DNA in HepG2.RL1 cells were >625 and >5556, respectively, by qPCR. It is noteworthy that the EC50 values of lamivudine and adefovir tested under our in vitro conditions were different from those obtained in other observations with stable transfected cell lines.34–37 The EC50 variations among different studies may be because of differences in the HBV genome and plasmids transfected (by site-directed mutagenesis or from clinical isolates), the time of action and the viral DNA quantification methods used for phenotypic assays. Therefore, the EC50 values of each drug determined against wild-type and mutant HBV viruses should only be compared in parallel experiments. However, the fold-resistance values are comparable with those found in previous studies,34,36 and confirmed that viruses containing these mutations show a remarkably decreased sensitivity to lamivudine but not to adefovir. Moreover, unlike transient transfection assays, stable cell line-based assays are less variable among experiments and thus suitable for high throughput drug screening.
NCC was selected from a series of 4-N-alkyl-substituted fluoronucleosides, which have been synthesized in order to address the toxicity issue of the parent compound FNC (2′-deoxy-2′-β-fluoro-4′-azido-β-d-arabinofuranosyl cytidine),38 and after in vivo evaluation, NCC exhibited a significantly improved toxicity profile in mice. The results from the MTT test showed that NCC inhibited the growth of HepG2.2.15 cells and HepG2.RL1 cells with CC50s of 725.81±23.34μM and 782.38±12.17μM, indicating that NCC causes less toxicity on cells and lower cytotoxicity than adefovir. The in vitro anti-HBV activity of NCC had been assessed in the HepG2.2.15 cells and HepG.CW cells in the previous study.15 In this study, we showed that NCC is a potent inhibitor of wild-type HBV in vitro. Moreover, the present study showed that NCC inhibited the replication of lamivudine-resistant HBV clinical isolates in a dose-dependent manner. In HepG2.RL1 cells, NCC inhibited HBeAg expression almost as efficiently as adefovir, while inhibited intracellular DNA replication more efficiently than adefovir (0.84±0.12μM versus 2.24±0.11μM). It is interesting that the EC50 value of NCC on extracellular DNA replication is still much lower than that of adefovir (0.46±0.09 versus 4.18±0.18μM), even though the resistance factor of NCC is a little higher than that of adefovir (3.29 versus 1.13). The promising activity of NCC against the lamivudine-resistant HBV may be associated with the novel chemical structure of NCC such as 3′-OH, 4′-azido and N-cyclopropyl, which do not share the same resistance profile as lamivudine. Furthermore, the in vitro therapeutic potency of a drug should take into account its antiviral capacity and cytotoxicity because a high therapeutic index (EC50/CC50) is preferable for a drug to have a favorable safety profile. In combination with the above results, the in vitro cross-resistance profile of NCC for the clinical lamivudine-resistant strain studied here was more favorable than those of adefovir.
ConclusionIn conclusion, the HepG2.RL1 cell line, established by introducing a clinical lamivudine-resistant HBV isolate with rtL180M/M204V mutations into HepG2 cells, was capable of stably producing HBV virions. The cell line should be helpful for screening antiviral agents active against lamivudine-resistant HBV and can streamline the screening protocol and directly evaluate the effects of new antiviral agents on clinical HBV strains. In addition, our studies indicate that NCC possesses antiviral activity against both wild-type and lamivudine-resistant HBV with low toxicity, and suggest that NCC deserves further development as a new and potential inhibitor of resistant HBV.
Conflicts of interestThe authors declare no conflicts of interest.
This work was supported by grants from the Natural Science Foundation of China (grant number 81302637), and the Young Innovation Foundation of The First Affiliated Hospital of Zhengzhou University of China (grant number 2015192).We appreciate Doctor Sheng-Ping Huang from Wuhan Institute of Virology, Chinese Academy of Sciences for kindly providing eukaryotic expression vector pcDNA3.1 (+). We thank Prof. Xiao-Ang Yang from Zhengzhou University for providing helpful advice and invaluable support for experiments.
