To investigate the genetic variation and molecular epidemiology characteristics of Human Respiratory Syncytial Virus (HRSV) in Guizhou Province, nasopharyngeal aspirates were collected from patients with acute respiratory infection (ARI) in Guizhou Provincial People's Hospital, from December 2017 to March 2018, and inoculated to Hep-2 cells to isolate HRSV. Cells that showed cytopathic effect (CPE) were then confirmed by indirect immunofluorescence assay and reverse transcription. The sequence of the PCR products was determined for HRSV isolates, and the genetic variation was analyzed. Out of 196 nasopharyngeal aspirate samples, HRSV were isolated in 39. The second hypervariable region at the 3′ terminal of glycoprotein gene (HVR2) sequence analysis showed that subgroup A was dominant. Seventy-nine percent of the isolates belonged to subgroup A, ON1 genotype, and 21 % belonged to subgroup B, BA9 genotype, which indicates that the dominant HRSV circulating in Guizhou Province was subgroup A, genotype ON1, co-circulating with a less prevalent subgroup B, genotype BA9.
Human Respiratory Syncytial Virus (HRSV) is one of the most important pathogens causing acute respiratory tract Infections (ARI) in infants, elderly, and immunocompromised people worldwide.1,2 HRSV infection is common, with more than 90 % of children infected before the age of two, with 0.5%–2% of cases requiring hospitalization due to severe symptoms.3 In 2015, Shi et al.4,5 estimated that 33.1 million cases of HRSV ARI occurred in children under five, including 3.2 million hospitalizations and 59,600 in-hospital deaths. The costs associated with the HRSV infection are high due to lack of a licensed vaccine and specific treatment.6
HRSV is a single-negative-strand RNA virus belonging to the Paramyxoviridae family genus pneumoviridae.7 The genomic RNA of HRSV has about 15,200 nucleotides in length containing 10 genes, which are NS1, NS2, N, P, M, SH, G, F, M2, and L, expressing 11 proteins. Among them, protein G is one of the important antigens against which the host often targets to produce protective antibodies. However, the virus can escape host's immune response through antigen mutation, which is an important cause of recurrent HRSV infection.8 HRSV is divided into two subtypes, A and B; variability among these subtypes is concentrated in two highly variable regions that is part of the extracellular domain of protein G. According to the sequence of a second hypervariable region (HVR2) within the G gene, the two groups of HRSV are further divided into genotypes.4,9 In recent years, the most common genotypes worldwide have been the ON1 genotype of subgroup A and the BA genotype of subgroup B.1,10–13
Guizhou Province is located in the southeastern part of southwest China, with jurisdiction over nine prefectures, among them three are the minority autonomous prefectures, and the capital city is Guiyang. Guizhou Province is a multi-ethnic province with a total of 56 ethnic groups with ethnic minorities accounting for 39 % of the province's total population. Some of the minorities live in isolated tribes. Hence it is important to study the molecular epidemic character and the evolution of HRSV in Guizhou Province. Some provinces in China have reported the genetic variation characteristics of HRSV, but Guizhou Province lacks such basic data.14–20 In order to understand the HRSV infection situation and the molecular epidemic characteristics of HRSV in Guizhou Province, we carried out HRSV isolation from patients with ARI aged less than five years old in Guiyang, from December 2017 to March 2018, and analyzed the HVR2 sequence of the G gene of the isolates. This study provides the basic data for the prevention and control of HRSV in Guizhou Province.
MethodsClinical samplesNasopharyngeal aspirates were collected from hospitalized children under five years old with ARI, mainly presenting with bronchiolitis or pneumonia, in Guizhou Provincial People's Hospital from December 2017 to March 2018. The study was approved by the Ethics Committees of Guizhou Provincial People's Hospital (approval number: 2017105).
Isolation of HRSVThe nasopharyngeal aspirates were treated with penicillin (100units/mL) and streptomycin (100μg/mL) before being inoculated to Hep-2 cells (ATCC) cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO, CA) supplemented with 2 % fetal bovine serum (FBS; GIBCO). The cytopathic effect (CPE) of inoculated Hep-2 cells was observed everyday for seven days. CPE negative samples at the seventh day after inoculation were blindly inoculated for two consecutive passages and CPE positive samples recorded and stored at −80°C for future use.
Indirect immunofluorescence assayCPE positive and uninfected (mock) Hep-2 cells were digested by 0.02 % EDTA, spotted to eight well glass slide (MP Biochemicals, CA, USA) separately. The slides were fixed with cold acetone after air drying the cells. After blocking with 3 % BSA for one hour at room temperature, the slides were incubated with sheep anti-HRSV IgG antibody (1:500, Millipore) at 37°C for one hour followed by washing three times with PBS, the slides were reacted with Alexa Fluor 488 labelled anti-sheep IgG (1:500, Invitrogen) at room temperature for one hour. After additional three washes with PBS, the slides were air dried, sealed with mounting medium for fluorescence (Vector Laboratories, Inc.). The samples were subsequently examined using a fluorescence microscope.
Reverse transcription, PCR and sequencing analysisTotal RNA was extracted from 500μL of the viral isolates culture fluid with Trizol (Invitrogen) according to the manufacturer’s instructions.21 Reverse transcription was performed at 25°C for 10min and 37°C for 60min, in a reaction mixture containing 11μL of RNA, 4μL of 5× First Strand Buffer (Invitrogen), 1mL of 100mM DTT, 1μL of 10mM dNTP Mix, 1μL of RNase inhibitor (TaKaRa), 1μL of random primers and 1μL of super script III (Invitrogen) in a final volume of 20μL.
Nested PCR was carried out with two sets of primers to amplify the three prime end of HRSV 10G gene for all the isolates. For the first amplification forward SH1 (5′ CACAGTKACTGACAAYAAAGGAGC) and reverse F164 (5′ GTTATGACACTGGTATACCAACC) primers were used, followed by a second amplification with ABG490 (5′ ATGATTWYCAYTTTGAAGTGTTC) and F9AB (5′ CAACTCCATKRTTATTTGCC) primer pair.6 One μL of the reverse transcription reaction mixture was used for the first amplification, whereas 1μL of the first amplification mixture was used for the second PCR. PCR reaction mixtures contained 10×LA PCR buffer, 1mM MgCL, 2.5mM of each dNTP Mix, 0.25μM of each primer and five units of LA Taq polymerase (TaKaRa). PCR conditions for the first amplification were: 94°C for two minutes, 34 cycles of: 94°C for 30s, 55°C for 30s and 72°C for one minute and 20s. One μL of the first amplification mixture was used for the second PCR, which was performed using the same reaction mixture at 55°C but with the second primer pair and shortened extension step of one minute. The amplification products were separated on a 1 % agarose gel.
Gel purified PCR products were used to set up the sequencing reactions using the ABG490 and F9AB primers the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Sequencing and sequence analysis were performed on a 3730 Genetic Analyzer (Thermo Fisher Scientific, USA).
Homology analysisThe obtained sequence alignments were performed using CLUSTALW. The nucleotide sequences of subgroup A and B viruses were confirmed by using BioEdit software (http://www.mbio.ncsu.edu/BioEdit/) and homology analysis was conducted using DNASTAR software.22
Phylogenetic analysisPhylogenetic trees of the G protein were performed using CLUSTALW. The nucleotide sequences of subgroup A and B viruses were confirmed using BioEdit software and homology analysis was done using DNASTAR.9 Bootstrap probabilities were calculated after 1000 iterations to evaluate confidence estimates and bootstrap values for viruses were confirmed using Bio Edit software. Homology between genotypes were estimated using the MEGA p-distance method, which calculates proportion (p) of nucleotide sites at which two sequences being compared are different.6
Analysis of deduced amino acid sequencesDeduced amino acid sequences were generated using ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) and the obtained amino acid sequences were analyzed using DNASTAR for homology analysis. BioEdit was used to compare the amino acid sequences of the HVR2 with the prototype strain.
ResultsHRSV isolation and identificationFrom December 2017 to March 2018, we collected 196 nasopharyngeal aspirates samples from patients with ARIs in Guizhou Province, for HRSV virus isolation by Hep-2 cell inoculation. HRSV positive isolates showed typical CPE. The infected cells became round and fused together forming giant cells (Fig. 1A and B). Out of the 196 samples, we obtained 39 (19.90 %) HRSV isolates. IFA results showed that all the isolates were reactive to anti-HRSV antibody (Fig. 1C and D). RT-PCR results confirmed that these isolates were positive for HRSV. The PCR products of these isolates were sent for sequencing and the results showed 31 strains (79 %) of subgroup A and eight strains (21 %) of subgroup B.
Homology analysis
The gene sequences obtained by sequencing the PCR products were confirmed with BioEdit and were compared with A2 and B prototype strains CH18537 by using Clustal W and BioEdit software. The sequence lengths obtained for subgroup A and subgroup B isolates were 516 nucleotides and 505 nucleotides, respectively. The homology analysis was performed using DNASTAR.
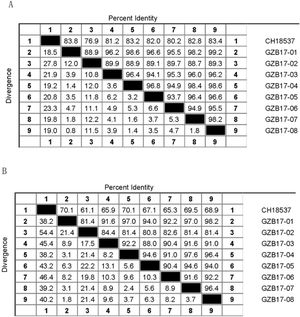
Between the prototypes strain A2 and the Guizhou subgroup A isolates, the rates of identity were 86.1%–87.9% at the nucleotide level and 79.6%–81.6% at the amino acid level. Identity within the Guizhou subgroup A isolates were 94.4%–100% and 87.7%–100% at the nucleotide level and at the amino acid level, respectively (Figs. 2A, B and S1). The rates of identity between the Guizhou B isolates and the prototypic genotype B strain CH18537 were 76.9%–83.8% at the nucleotide and 61.1%–70.1% at the amino acid level. Identity within the Guizhou subgroup B isolates were 88.7%–99.2% and 80.8%–98.2% at the nucleotide level and at the amino acid level, respectively (Figs. 3A, B and S2).

Comparison of homology between the Guizhou HRSV subgroup A isolates 3′ end of G gene nucleotide and amino acid sequences with that of the prototype strain A2 in colored heat map. A, comparison of nucleotide sequences homology of subgroup A; B, homology comparison of amino acid sequences of subgroup A. Red indicates the maximum identity, blue indicates the minimum identity, white indicates the middle value, green indicates the maximum divergence, red indicates the minimum divergence, yellow indicates the middle value.

Comparison of homology between Guizhou HRSV subgroup B isolates 3′ end of G gene nucleotide and amino acid sequences with that of the prototype strain CH18537 in colored heat map. A, comparison of nucleotide sequences homology of subgroup B; B, homology comparison of amino acid sequences of subgroup B. Red indicates the maximum identity, blue indicates the minimum identity, white indicates the middle value, green indicates the maximum divergence, red indicates the minimum divergence, yellow indicates the middle value.
The sequences of the HVR2 G genes from 31 subgroup A and eight subgroup B isolates obtained in this study and some representative genotypes sequences downloaded from GenBank were used for phylogenetic analysis.
The newly isolated 31 HRSV subgroup A strains were discovered bearing a 72-nt duplication when compared the sequence of the prototype virus (reference strain M11486). These results revealed that all 31 subgroup A viruses were clustered into genotype ON1, which were most closely related to ON1 strains KX533701 KX533692, KX533588, KT765058, JN257693, and KC731482 from China (Beijing, Sichuan), Canada and India, respectively (Fig. 4A).
 and subgroup B (B) isolates. Trees were generated by using the Neighbor-joining methods. Bootstrap values greater than 70 % are shown at the branch notes. The value represents the bootstrap probability. The genotypes obtained by isolates in this study are indicated by brackets on the right side. Reference sequences describing each genotype (ON1, NA1 to NA4, GA1 to GA7, SAA1, GB1 to GB4, SAB1 to SAB3, BA1 to BA10) were obtained from the Genbank database and included in the tree. Prototype strains (strain A2 for subgroup A and strain CH18537 for subgroup B) were used as the outgroup sequences.")
Phylogenetic trees of nucleotide sequences from the second variable region of the G gene of HRSV subgroup A (A) and subgroup B (B) isolates. Trees were generated by using the Neighbor-joining methods. Bootstrap values greater than 70 % are shown at the branch notes. The value represents the bootstrap probability. The genotypes obtained by isolates in this study are indicated by brackets on the right side. Reference sequences describing each genotype (ON1, NA1 to NA4, GA1 to GA7, SAA1, GB1 to GB4, SAB1 to SAB3, BA1 to BA10) were obtained from the Genbank database and included in the tree. Prototype strains (strain A2 for subgroup A and strain CH18537 for subgroup B) were used as the outgroup sequences.
All newly HRSV subgroup B isolates were found to cluster in the BA genotype and had the signature 60 bp duplication in the G protein when aligned with prototype B reference strain CH18537. The HRSV subgroup B isolates were most closely related to BA9 strains KT781400, KJ658809, GU550503, KT765101, and HM459879 from China (Hunan, Shanghai, Sichuan and Chongqing) and Japan, respectively (Fig. 4B).
Analysis of deduced amino acidsThe predicted amino acid sequences of HVR2 G gene sequences deduced from the 270 nucleotide fragment of the subgroup A and subgroup B isolates were used to generate a consensus sequence at amino acid level for each genotype in comparison with prototype strains A2 and CH18537, respectively. When compared with A2 strain, the newly isolated HRSV subgroup A strains were ON1 genotype with the insertion of 24 amino acids in HVR2 region (Fig. 5A). The newly isolated subgroup B strains obtained in this study had a duplication of 20 amino acids in HVR2 region when compared with CH18537 strain (Fig. 5B).
; B, deduced amino acid alignment of HVR2 from Guizhou subgroup B isolates shown relative to the sequence of strain CH18537 (Number acc. nº JX198143). Different amino acids are shown in different colors, identical residues are indicated by dots, dashes designate gaps corresponding to nucleotide insertions and asterisks indicate stop codons.")
Deduced amino acid alignment of HVR2 from Guizhou HRSV isolates shown relative to the sequence of prototype strains. A, deduced amino acid alignment of HVR2 from Guizhou subgroup A isolates shown relative to the sequence of strain A2 (acc. nº M11486); B, deduced amino acid alignment of HVR2 from Guizhou subgroup B isolates shown relative to the sequence of strain CH18537 (Number acc. nº JX198143). Different amino acids are shown in different colors, identical residues are indicated by dots, dashes designate gaps corresponding to nucleotide insertions and asterisks indicate stop codons.
To investigate HRSV molecular epidemic characteristics in Guizhou Province, we isolated 39 HRSV strains from ARI patients in Guizhou Province from December 2017 to March 2018. These isolates were confirmed to be HRSV by IFA and RT-PCR. The genotypes of these isolates were identified by sequence analysis. This is the first report of HRSV virus isolation and genotyping in Guizhou Province, China.
The virus isolation rate was lower in our study (19.9 %) compared to others.9,10,23 The main reason may be because the nasopharyngeal aspirate sample used for virus isolation was the remaining from clinical bacteria culture tests. The amount left for virus isolation was sometimes insufficient. Another reason may be because we used only one cell line (Hep-2) for virus isolation. To improve the virus isolation rate in future studies, using several cell lines for virus culture and fresh clinical samples maybe necessary. Compared with the studies of direct PCR amplification using extracted RNA from nasopharyngeal samples without virus isolation, the positive rate of direct PCR was much higher, indicating that gene detection methods were much more sensitive.24,25 Russo M et al.24 reported that out of 520 samples, 238 were positive by real time RT-PCR (positive rate 55.5 %). The positive rate of HRSV was 46.11 % in 180 clinical samples using RT-PCR in another report.25
The epidemic pattern of HRSV subtype varies from region to region. In a given year, subtype A and subtype B may co-exist in a region, or one of them may be dominant, although subgroup A infection is dominant in most parts of the world.26 For example, in the six consecutive epidemic seasons from 2000 to 2006 in Beijing area, subgroup A dominated in 2001–2004, subgroup B dominated in 2004–2005, and co-dominance by two subgroups in the 2005–2006 epidemic season.27 A total 5483 children with respiratory diseases were tested in Guangzhou city for three years. Both subgroup A positive cases (51.2 %, 373/729) and subgroup B positive cases (48.8 %, 356/729) were found. Subgroup B was dominant in 2013, subgroup A was dominant in 2015, and subgroup A and subgroup B were transmitted together in 2014.28 In 2010–2014, there were 570 cases of HRSV infection in Mexico, of which 407 cases belonged to subgroup A, and only 27.54 % were subgroup B6; In Croatia, between 2011 and 2014, subgroup A was the dominant type, accounting for 76 % of HRSV infections.29
Between 2011 and 2013, subgroup A was reported to be dominant in southern Brazil, out of 167 detected HRSV cases, 136 were subgroup A.11 Similarly, in the period 2013–2015, subgroup A accounted for 76.7 % of the 73 cases of HRSV infection in Sao Paulo, Brazil.30
In this study, out of the total 39 HRSV isolates, 31 were subgroup A and eight were subgroup B. Indicating that both subgroups were co-circulating in Guizhou Province, but subgroup A predominated as seen in other areas of the world.
Genotype shifting of HRSV is common. From 2003–2008, studies have shown that the dominant genotype of subgroup A was GA2 in some provinces of China while the dominant genotype of GA2 was replaced by genotype NA1. NA1 genotype was first reported in Beijing from 2008 to 2013.31 Similarly, the ON1 genotype was first found in Canada in 2010 and has since been reported in the United States, Latvia, South Korea, Thailand, Japan, Kenya, West India, and other countries.32 In China, the ON1 genotype was first detected in Shanghai in February 2011.33 Since then, ON1 genotypes have become increasingly common, replacing NA1 genotypes as the dominant genotype in China.31,33 In Brazil, the new HRSV-A genotype ON1was introduced in 2012 and became prevalent in 2013.11 Globally, different ON1 genotypes have been reported in 21 countries and have rapidly spread around the world.32 As for subgroup B, the BA genotype was first reported in Trento in 1999, then detected in Japan in 2003, and subsequently in South Africa, China, Brazil, and Thailand, becoming the dominant subgroup B genotype worldwide.34 In China, the BA strain was first identified in Beijing in 2004 and rapidly became the dominant one.22
Although ON1 and BA strains appear to have been more successful in replacing other genotypic strains, the reason for their dominance is not clear. Some authors reported that there is no indication that ON1 and BA strains lead to more serious disease,6,22 while others reported that ON1 genotype showed an increased tendency to develop severe disease.11,35 Among the 72 nucleotide duplication in ON1 genotype of subgroup A and the 60 nucleotide duplication in BA9 genotype of subgroup B, some minor substitutions at the nucleotide level and amino acid level were found. Whether these minor mutations cause the diversity reported by different research groups is not clear. More detailed studies focusing on the virology and immunology characteristics of different genotypes are needed to illustrate the mechanism of genotype shift and the pathogenicity of the virus.
Phylogenetic analysis revealed that all isolates of subgroup A in Guizhou Province were classified as ON1 with a 72 nucleotide duplication in the HVR2 gene, while all subgroup B isolates were clustered to BA9 with typical duplication of 60 nucleotides in the HVR2 domain. This finding indicates that ON1 genotype in subgroup A and BA9 genotype in subgroup B were predominant in Guizhou Province as seen in other places.
In conclusion, we reported for the first time the isolation and identification of HRSV in Guizhou Province. The 3′ end of G gene of the 39 isolates was sequenced and analyzed. In Guizhou Province, both subgroup A and B were circulating in 2017–2018, but the majority of clinical isolates was subgroup A, ON1 genotype. All subgroup B isolates were of BA9 genotype. Further surveillance of HRSV genetic variation is needed for preventing and controlling of HRSV infection in Guizhou Province.
Author contributionsFY, YC, ZY and YN designed the study. TZ, BW, WH, LL, ZL, BY, QX, KC, FH, HZ and FY performed the experiments. TZ, ZY and FY analyzed the data. TZ and FY wrote the paper. All authors read and approved the final manuscript.
FundingThis study was supported by Fund of the Science and Technology Department of Guizhou Province ([2018]2785, [2015]4015, [2017]5781, [2018]5706, [2018]5801), the National Natural Science Foundation of China(2018)81860003, (2019)81960001.
Ethics approval and consent to participateThe study was approved by the Ethics Committees of the China Guizhou Provincial people's Hospital (Approve number 2017105).
Conflict of interestThe authors declare no conflicts of interest.
The authors wish to thank the staff of Guizhou Provincial People's Hospital for the assistance of sample collection and research cooperation. We thank Dr. Klarke M. Sample from the Guizhou Provincial People’s Hospital for editing the English of our manuscript.